Pathologie de la cornée
Coordonné par C. Burillon
M. Beylerian, G. Ho Wang Yin, L. Hoffart
Les dystrophies de cornée chez l’enfant englobent un groupe hétérogène de maladies génétiques bilatérales non inflammatoires qui sont en général limitées à la cornée. Cette terminologie est imprécise et il existe de nombreuses controverses en raison des multiples définitions phénotypiques rapportées par de nombreux auteurs.
La prévalence de ces pathologies n’est pas précisément connue du fait de leur rareté. Elle est variable en fonction du type de dystrophie. L’âge de début varie en fonction du type de dystrophie. Si certaines dystrophies s’expriment dès l’enfance, rares sont celles ayant un retentissement visuel avant l’âge adulte et rares sont celles qui nécessitent une kératoplastie, encore moins avant l’âge adulte. Le risque d’amblyopie est alors à confronter à celui lié à la kératoplastie transfixiante chez l’enfant. La dystrophie héréditaire endothéliale congénitale (congenital hereditary endothelial dystrophy [CHED]) est la plus fréquente et de meilleur pronostic après kératoplastie par rapport aux autres dystrophies.
Dans ce sous-chapitre, nous analyserons la définition des dystrophies cornéennes, leurs classifications, la description clinique des différents types selon la mutation génétique impliquée et leurs caractéristiques phénotypiques, et enfin la prise en charge thérapeutique qui peut être proposée.
Le terme dystrophie vient des mots grecs : dys- (du grec ancien δυσ-, dus- qui exprime une idée de difficulté, de mauvais état) et -trophie (du grec ancien τροφή, trophê : nourriture, croissance ; suffixe des mots relatifs à la croissance, au développement d’un organisme).
Dans ce sous-chapitre, nous analyserons la définition des dystrophies cornéennes, leurs classifications, la description clinique des différents types selon la mutation génétique impliquée et leurs caractéristiques phénotypiques, et enfin la prise en charge thérapeutique qui peut être proposée.
Il n’y a pas de définition universelle pour le terme de dystrophie. Les manifestations cliniques phénotypiques des dystrophies de cornée chez l’enfant varient grandement selon l’entité. Ce terme est utilisé depuis 150 ans pour définir un groupe d’entités pathologiques dont l’origine n’est ni traumatique, ni infectieuse et qui résulte d’un manque d’innervation ou de nutrition. Plusieurs articles ont récemment démontré une implication génétique dans la genèse de ces atteintes.
Les dystrophies cornéennes correspondent généralement à un groupe de pathologies héréditaires souvent bilatérales, symétriques, lentement progressives et non liées à des facteurs environnementaux ou des maladies systémiques.
Néanmoins, certaines dystrophies cornéennes chez l’enfant peuvent être cliniquement unilatérales, telle que la dystrophie postérieure polymorphe ; des anomalies biologiques peuvent également être associées comme dans la dystrophie de Schnyder où l’hypercholestérolémie est fréquente.
Cliniquement, une dystrophie cornéenne doit être suspectée lors d’une perte progressive de la transparence cornéenne ou si des opacités cornéennes apparaissent spontanément chez des enfants avec des antécédents familiaux ou une notion de consanguinité. Le diagnostic clinique repose sur l’âge de début et sur l’apparence à l’examen biomicroscopique. Les explorations paracliniques peuvent aider au diagnostic avec éventuellement un examen histologique en microscopie optique et microscopie électronique de transmission après biopsie cornéenne. Plus récemment développée, la microscopie confocale apporte une aide précieuse au diagnostic, l’aspect des dystrophies cornéennes étant désormais bien codifié en microscopie confocale pour la plupart d’entre elles [1, 2]. La biologie moléculaire permet de confirmer le type de dystrophie lorsque la mutation génétique a été identifiée.
L’introduction de génotypage a révolutionné notre connaissance des dystrophies cornéennes. Le génotypage révèle l’hétérogénéité génotypique qui caractérise une dystrophie – telle la dystrophie de Meesmann pouvant être associée à des gènes différents (KRT3 et KRT12) – ou l’hétérogénéité phénotypique du gène TGFB1 qui est associé à de nombreux phénotypes différents – telles la dystrophie de Reis-Bücklers ou la dystrophie granulaire de type 1 et 2.
La classification internationale des dystrophies cornéennes proposée en 2008 par l’International Committee for classification of corneal dystrophies (IC3D) [3] reposait sur la classification anatomique traditionnelle organisée selon la couche cornéenne impliquée. Une classification plus récente en catégories fondée sur le niveau de preuve (connaissances cliniques, pathologiques et génétiques) a été suggérée par G.-K. Klintworth. Ainsi, les dystrophies de cornée peuvent être classées selon l’aspect clinique et selon les gènes impliqués en quatre catégories (tableau 9-1).
Tableau 9-1 – Classification des dystrophies cornéennes fondées sur différents niveaux de preuves (IC3D).

En 2015, la classification IC3D a été revisitée et l’IC3D-2 a été proposée [3] car le fait de ne se fonder que sur un niveau anatomique (par couche spécifique) présente certaines limites ; les dystrophies cornéennes sont désormais subdivisées en : dystrophies épithéliales et subépithéliales, dystrophies épithéliales stromales liées au TGFB1, dystrophies stromales et dystrophies endothéliales (tableau 9-2).
Cependant, le diagnostic d’une dystrophie cornéenne pédiatrique reste un défi. La plupart des données publiées concernent des cas diagnostiqués à l’âge adulte et peu d’iconographie permet de documenter les atteintes des dystrophies congénitales et/ ou néonatales et leur évolution.
Dans la classification des dystrophies de cornée IC3D-2, on peut isoler les dystrophies congénitales et/ou néonatales qui apparaissent dans l’enfance (tableaux 9-3 à 9-6).
On note que la plupart des dystrophies cornéennes ne sont pas associées à des atteintes systémiques et se traduisent par la survenue d’opacités cornéennes différentes qui expliquent le retentissement variable sur l’acuité visuelle.
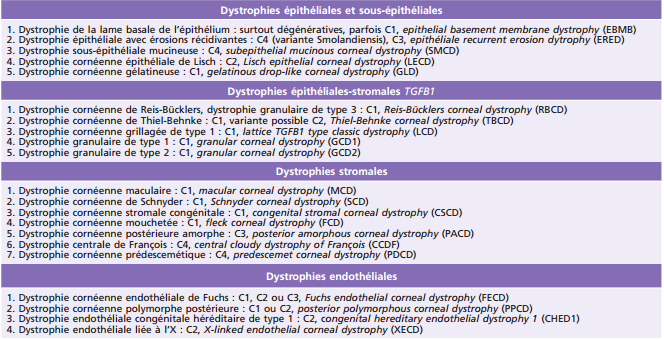
Tableau 9-3 – Dystrophies cornéennes épithéliales et sous-épithéliales chez l’enfant.
AD : autosomique dominante.
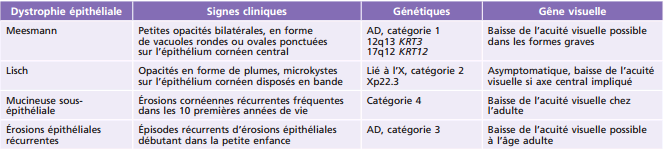
Tableau 9-4 – Dystrophies cornéennes épithéliales-stromales TGFB1 chez l’enfant.
AD : autosomique dominante.
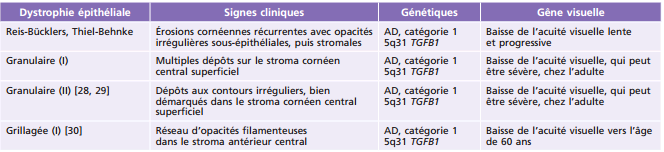
Tableau 9-5 – Dystrophies cornéennes stromales chez l’enfant.

AD : autosomique dominante ; AR : autosomique récessive.
Tableau 9-6 – Dystrophies cornéennes endothéliales chez l’enfant.
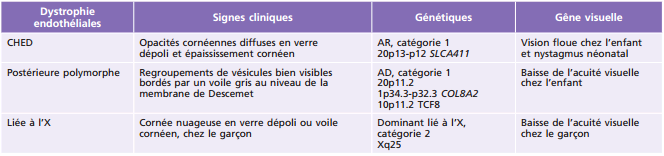
AD : autosomique dominante ; AR : autosomique récessive.
Le diagnostic d’une dystrophie de cornée chez l’enfant est délicat à établir, les manifestations cliniques étant le plus souvent partielles. Dans la littérature, de nombreux auteurs évoquent la nécessité de se fonder sur une corrélation phénotype-génotype pour établir un diagnostic [4] et non plus seulement sur une évaluation clinique qui peut être source d’erreur et conduire à la description de nouvelles entités de dystrophies (de catégorie 4) lorsqu’un phénotype n’est pas évocateur d’une dystrophie décrite.
Deux approches non exclusives permettent d’identifier des gènes responsables de maladies héréditaires [5] : l’approche par gène candidat (dite génétique « classique » ) et l’approche par génétique positionnelle ou cartographie génétique (génétique « inverse » ).
De nombreuses pathologies oculaires ont bénéficié de l’approche par cartographie génétique fondée sur les études de liaison génétique (ou étude de linkage) et de cartographie par homozygotie par filiation (dans des familles consanguines, dans la plupart des cas, en recherchant des zones d’homozygotie sur le génome). Elles sont fondées sur la recherche d’une coségrégation d’un marqueur chromosomique avec une pathologie donnée dans une ou plusieurs familles bien caractérisées sur le plan clinique. Ces études permettent de montrer que le gène muté se situe dans une région chromosomique donnée. Les marqueurs génétiques utilisés classiquement sont des microsatellites constitués de répétitions courtes d’acide désoxyribonucléique (ADN) réparties à travers tout le génome et plus récemment l’approche par puces single nucleotide polymorphism (SNP). La localisation chromosomique est alors précisée et il est possible ensuite d’identifier le gène qui sera cloné et séquencé, en utilisant notamment des chromosomes artificiels de levure, des cosmides ou des banques d’ADN tissus spécifiques. Une fois que la mutation dans un gène est identifiée, il s’agit ensuite de relier celle-ci à un mécanisme pathogénique sous-jacent (un modèle animal est alors souvent nécessaire) et d’établir une corrélation entre le phénotype et le génotype grâce à une description sémiologique précise du clinicien.
Les modes de transmission des dystrophies cornéennes sont multiples (autosomique récessif, autosomique dominant, lié à l’X) et il existe une grande hétérogénéité génétique de ce groupe d’affections. Plus d’une dizaine de gènes sont actuellement impliqués dans les dystrophies cornéennes.
Parmi les dystrophies héréditaires, certaines ont été localisées par des études de liaison génétique sur le chromosome 5 en 5q31 [6] : dystrophie cornéenne de Reis-Bücklers (Reis-Bücklers corneal dystrophy [RBCD]), dystrophie granulaire de type 3 (RBCD), dystrophie cornéenne de Thiel-Behnke, dystrophie cornéenne grillagée de type 1 (lattice corneal dystrophy [LCD]), dystrophie granulaire de type 1 (granular corneal dystrophy type 1 [GCD1]) et dystrophie granulaire de type 2 (GCD2). Le gène responsable de ces affections s’est révélé être le TGFB1 (ou kérato-épithéline). L’origine moléculaire de plusieurs formes différentes de dystrophies cornéennes peut être commune, montrant ici l’exemple de variabilité allélique : des mutations différentes dans un même gène codant pour une protéine cornéenne sont responsables de dystrophies cornéennes cliniquement différentes.
En conséquence, la génétique moléculaire est un outil majeur qui est en train de révolutionner le diagnostic et le traitement des maladies oculaires héréditaires [7].
En 2003, aux États-Unis, le National Eye Institute relevant du National Institute of Health (NIH) a créé un réseau national de génotypage et de phénotypage des maladies ophtalmiques : eyeGENE est une banque d’ADN où l’information génétique moléculaire des patients atteints de maladies oculaires héréditaires peut être couplée à une description phénotypique. Grâce à eyeGENE, les professionnels de santé peuvent obtenir des résultats génétiques certifiés pour leurs patients.
La MECD est due à une mutation autosomique dominante sur le locus 12q13.13 où se situe le gène kératine K3 (KRT3) ou sur le locus 17q11-q1 où se situe le gène kératine K12 (KRT12), variante de la dystrophie de Stocker-Holt, causée par un changement de l’acide aminé p. Arg19Leu sur la cytokératine 12, classant cette dystrophie dans la catégorie 1 [8].
Les lésions se développent dès la petite enfance. La MECD reste souvent asymptomatique jusqu’à l’âge moyen, les symptômes qui peuvent ensuite apparaître sont une photophobie, une baisse de l’acuité visuelle légère et transitoire et un astigmatisme irrégulier. Les symptômes de la forme variante de la dystrophie de Stocker- Holt sont généralement plus sévères et plus précoces [9].
Chez les enfants, présence de minuscules vésicules intra-épithéliales, souvent centrales. La microscopie optique révèle des kystes intra-épithéliaux et un épithélium parfois épais et désorganisé. Dans 85 % des cas, les kystes sont diffus sur toute la surface épithéliale. La coalescence de plusieurs kystes peut entraîner des opacités linéaires et la sensibilité de la cornée peut être réduite.
La totalité de la cornée montre des opacités épithéliales punctiformes grisâtres qui se colorent avec la fluorescéine et des opacités linéaires fines qui peuvent apparaître sous forme de cornea verticillata.
Les microkystes intra-épithéliaux apparaissent sous forme de matériaux hyperréflectifs dans les couches superficielles de la cornée, avec une ligne de démarcation visible entre les microkystes et les cellules épithéliales normales, correspondant typiquement à la démarcation visible entre les zones claires de cornée et les zones de cornée affectées [10].
La maladie persiste au cours de la vie, l’évolution est souvent stationnaire ou lentement progressive. Dans les cas graves, des cicatrices sous-épithéliales entraînent une opacification grisâtre sur la cornée centrale.
La transmission est dominante liée a l’X. Le gène impliqué a été localisé sur le bras court du chromosome X (Xp22.3), classant cette dystrophie dans la catégorie 2 [11].
Les lésions se développent généralement dans l’enfance. Les symptômes se manifestent par une vision floue ou asymptomatique si l’axe pupillaire n’est pas impliqué.
Atteinte épithéliale : il s’agit d’opacités grises se présentant sous la forme de cornea verticillata avec une disposition radiale ou sous la forme de bandes, de flammes ou encore de plumes. En illumination indirecte, on note la présence de multiples kystes clairs.
Il y a quatre caractéristiques des cellules épithéliales anormales : cytoplasme hautement hyperréflectif et noyaux hyporéflectifs ; implication uniforme de toutes les couches épithéliales dans les zones affectées ; frontières nettes avec l’épithélium normal adjacent ; implication de la zone limbique [12].
L’évolution est très progressive.
La maladie n’a été rapportée que dans une seule famille.
L’étiologie est inconnue, le gène et le locus impliqués n’ont pas encore été découverts. Le mode autosomique dominant est probable mais une hérédité liée à l’X n’est pas exclue, classant cette dystrophie dans la catégorie 4.
Les épisodes douloureux d’érosions cornéennes récurrentes ont lieu au cours des 10 premières années de vie puis diminuent à l’adolescence.
Atteinte épithéliale puis sous-épithéliale : plus tard, les patients développent des opacités sous-épithéliales et un voile cornéen, surtout au centre de la cornée.
La SMCD progresse ensuite avec le temps, entraînant des opacités cornéennes et une perte de la vision.
La transmission est autosomique dominante, le locus et le gène sont encore inconnus classant cette dystrophie dans la catégorie 3.
Cette dystrophie est rare et les symptômes débutent en général vers l’âge de 4 ans par une photophobie ou par des douleurs oculaires, d’horaire inflammatoire, témoin d’érosions épithéliales et récurrentes. Les érosions sont d’apparition spontanée ou bien précipitées par un traumatisme, une exposition aux rayons ultraviolets (UV), à la poussière ou à la fumée.
- Atteinte épithéliale : les érosions épithéliales durent en général une semaine et sont ensuite accompagnées de périodes de rémission, sans signe clinique à la lampe à fente.
- Atteinte sous-épithéliale : au fur et à mesure des récidives, peuvent apparaître des opacités sous-épithéliales, avec une fibrose sous-épithéliale ou des nodules de type chéloïdes [13].
L’intensité et la fréquence des érosions épithéliales tendent à diminuer avec le temps et sont de moins en moins fréquentes vers l’âge de 30-40 ans.
Une baisse de l’acuité visuelle pourra apparaître secondairement, à l’âge adulte, en raison de l’opacification cornéenne centrale.
Transmission autosomique dominante de la mutation R124L du gène TGFB1 au niveau du locus 5q31 classant cette dystrophie dans la catégorie 1.
L’atteinte débute dans l’enfance par une baisse de l’acuité visuelle ou par des kératalgies secondaires à des ulcérations cornéennes.
- Atteinte sous-épithéliale et stromale antérieure : présence d’opacités géographiques irrégulières confluentes, de densités variables, sous-épithéliales qui se développent au niveau de la couche de Bowman et du stroma superficiel.
- Atteinte stromale profonde : ces opacités s’étendent ensuite vers le limbe et le stroma profond.
La RCBD peut être confondue avec la dystrophie de Thiel- Behnke (Thiel-Behnke corneal dystrophy [TBCD]), mais dans les stades précoces, la RBCD présente des opacités diffuses plus irrégulières avec des zones de cornée claires, alors que la TBCD présente plusieurs mouchetures avec un aspect en rayon de miel (formation réticulaire).
Présence d’une couche confluente et homogène de dépôts hyperréflectifs à bords antérieurs dentelés au niveau de la couche de Bowman et du stroma antérieur. Cette couche est plus épaisse au centre (72-132 mm) et devient plus mince en moyenne périphérie, pour enfin disparaître au limbe.
On observe des dépôts sous-épithéliaux, granulaires ou amorphes (sans ombres) et de réflectivité élevée. Dans la couche de Bowman, les dépôts sont irréguliers et hautement réfléchissants (plus réfléchissants que dans la TBCD). Enfin, des dépôts ronds ou en forme de fuseau peuvent être notés dans la partie antérieure et, dans de plus rares cas, dans le stroma postérieur.
Détérioration lentement progressive de la vision. La survenue des érosions cornéennes récidivantes a tendance à diminuer avec le temps.
Transmission autosomique dominante de la mutation R555Q du gène TGFB1 au niveau du locus 5q31 classant cette dystrophie dans la catégorie 1. Une autre mutation sur le chromosome 10q24 est possible (catégorie 2).
Les symptômes débutent dans la petite enfance par des douleurs, comme la RBCD, témoin d’érosions cornéennes récurrentes.
- Atteinte de la membrane de Bowman : les premiers signes sont des mouchetures isolées ou des opacités irrégulières dispersées au niveau de la couche de Bowman.
- Atteinte sous-épithéliale : ces opacités sont suivies par l’apparition d’opacités alvéolaires en rayon de miel sous-épithéliales symétriques, sans atteinte de la cornée périphérique.
- Atteinte stromale : avec le temps, les opacités peuvent évoluer vers le stroma et la périphérie de la cornée.
Les mêmes caractéristiques que dans la RBCD sont retrouvées, la principale différence réside dans le fait que dans la couche de Bowman, les opacités irrégulières sont moins réfléchissantes que dans la RBCD.
La baisse de l’acuité visuelle sera plus tardive que dans la RBCD.
Transmission autosomique dominante de la mutation R124C au niveau du locus 5q31 du gène TGFB1.
Les symptômes peuvent apparaître dès 3 ans, dans la plupart des cas au cours des 10 premières années de vie et se manifestent essentiellement par une photophobie ou plus rarement par des kératalgies, témoins d’érosions cornéennes récurrentes.
- Atteinte de la membrane de Bowman : on note des opacités granulaires brunâtres qui se développent de manière superficielle au niveau de la couche de Bowman. En rétro-illumination, ces opacités granulaires sont composées de vacuoles translucides associées à des grains de taille réduite, donnant un aspect de « chapelure écrasée » .
- Atteinte stromale : les opacités stromales centrales ne se prolongent pas jusqu’au limbe. La taille et le nombre de ces opacités granulaires augmentent avec l’âge des enfants, donnant un aspect de flocon de neige fig. 9-1. De plus, avec l’âge, les granules atteignent le stroma profond jusqu’à la membrane de Descemet. Les enfants homozygotes présentent des manifestations plus sévères.
L’apparence des dépôts granuleux dans la GDC1 est différente de ceux de la GCD2 où on note des dépôts d’amylose associés.
Présence d’opacités hyperréflectives en forme de flocons de neige ou de forme trapézoïdale observée.
Baisse de l’acuité visuelle, au fil des années, témoin de la confluence des opacités au niveau du stroma.
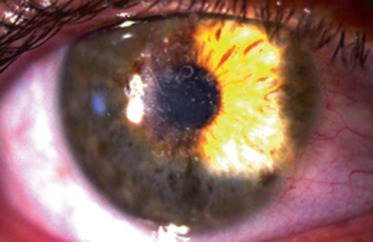
Fig. 9-1 Dystrophie granulaire type 1 : opacités stromales centrales n’atteignant pas le limbe chez un enfant de 16 ans.
Transmission autosomique dominante du gène TGFB1 au niveau du locus 5q31.
Les signes fonctionnels peuvent apparaître vers l’âge de 3 ans chez les patients homozygotes et vers 8 ans chez les hétérozygotes. Les manifestations sont essentiellement des kératalgies associées à des érosions épithéliales et une baisse de l’acuité visuelle qui évolue avec l’âge.
Atteinte stromale : les premiers signes sont discrets, il s’agit de petites opacités blanchâtres et superficielles, au niveau du stroma, qui peuvent être disposées de façon linéaire comme un collier de perles. Plus tard, ces opacités superficielles évoluent sous une forme ronde avec un centre mité, en forme d’anneau. La plupart des patients développent également des dépôts, hérissés, en forme d’étoile au niveau du stroma moyen. Les opacités de la phase finale sont translucides sous la forme de dépôts linéaires en forme de courts tirets, dans le stroma antérieur puis postérieur [16]. Les patients atteints de GCD2 présentent moins d’opacités stromales que ceux atteints de GCD1.
On observe la présence de dépôts réfléchissants ronds avec des bords bien délimités ou des dépôts trapézoïdaux irréguliers hautement réfléchissants dans le stroma antérieur (similaires à GCD1). Des dépôts linéaires et des ramifications avec des changements de réflectivité sont aussi observés (semblables à celles des dystrophies grillagées de type 1).
Lentement progressive avec présence, pour les homozygotes à l’âge adulte, de larges opacités denses, de formes irrégulières dans le stroma profond.
La transmission est autosomique dominante et implique le locus 5q31 du gène TGFB1 (mutation R124C) classant cette dystrophie dans la catégorie 1 [17]. C’est l’une des dystrophies les plus fréquentes rapportée en Occident.
Les symptômes se développent de façon bilatérale et asymétrique après l’âge de 10 ans et débutent par un gène oculaire ou une douleur oculaire, témoin d’érosions épithéliales.
- Atteinte épithéliale : les érosions cornéennes récurrentes peuvent précéder l’apparition des opacités cornéennes.
- Atteinte stromale : les premiers signes sont des opacités superficielles centrales, rondes ou ovoïdes (fleck-like), sous la forme de filaments en réseau opaque et linéaire, qui se développent dans le stroma antérieur central initialement, alors que la cornée périphérique reste transparente. Un haze stromal, central et paracentral, sous-épithélial, en verre dépoli peut apparaître en même temps que les filaments entrecroisés (fig. 9-2a, b).
Les structures linéaires du stroma se présentent sous un changement de réflectivité linéaire avec des marges mal délimitées. Ces lignes doivent être différenciées des autres images similaires, comme les champignons filamenteux.
La LCD1 est de progression lente et cause généralement une baisse de l’acuité visuelle que bien plus tard (60 ans).

Fig. 9-2 Dystrophie grillagée type 1.
a. Opacités fleck-like sous forme de filaments avec haze stromal chez un enfant de 15 ans. b. Réseau de filaments entrecroisés dans le stroma antérieur en rétro-illumination.
(Remerciements au Pr C. Burillon.)
Il s’agit d’une forme rare et sévère de dystrophie de transmission autosomique récessive, concernant le gène CHST6 sur le locus 16q22, classant cette dystrophie dans la catégorie 1 [18].
Les premiers symptômes apparaissent dans l’enfance et cette dystrophie peut s’accompagner d’une baisse sévère de l’acuité visuelle entre 10 et 30 ans.
- Atteinte stromale : dans un premier temps, des opacités centrales, superficielles, irrégulières et blanchâtres (fleck-like) se développent dans le stroma profond jusqu’à la membrane de Descemet et en périphérie au niveau du limbe. Simultanément, un haze progressif se développe, impliquant l’ensemble du stroma cornéen et responsable de la grave détérioration visuelle (fig. 9-3).
- Atteinte épithéliale : parfois des érosions épithéliales peuvent être associées.
- Atteinte membrane de Descemet : le stroma cornéen est aminci et, au fur et à mesure que la maladie progresse, des excroissances en forme de gouttes peuvent apparaître au niveau de la Descemet.
On peut observer des accumulations circonscrites et floues de dépôts hyperréfléchissants dans l’épithélium basal et le stroma.
Baisse de l’acuité visuelle chez l’adulte.
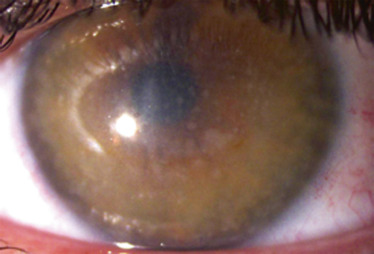
Fig. 9-3 Dystrophie maculaire : opacités centrales stromales irrégulières chez un enfant de 15 ans.
Transmission autosomique dominante du gène UBIAD1 du locus 1p36 classant cette dystrophie dans la catégorie 1.
Les symptômes apparaissent en général dès l’enfance mais le diagnostic est souvent retardé et réalisé entre 20 et 30 ans, chez des patients qui rapportent un éblouissement qui augmente avec l’âge, une baisse de l’acuité visuelle progressive. Le diagnostic peut être encore retardé chez les patients qui présentent une forme acristalline de la maladie (50 % ).
Les modifications cornéennes sont prévisibles en fonction de l’âge du patient.
Atteinte stromale : les enfants développent vers l’âge de 23 ans une opacité cornéenne, jaunâtre, centrale, annulaire ou en forme de disque, composée de cristaux sous-épithéliaux dans le stroma antérieur (fig. 9-4).
L’épithélium, la membrane de Descemet et l’endothélium ne sont pas touchés.
Entre 23 et 38 ans, un gérontoxon se développe et après l’âge de 38 ans, un haze stromal apparaît.
Des troubles systémiques associés ont été régulièrement rapportés : hypercholestérolémie IIA, III ou IV et genu valgum [19].

Fig. 9-4 Dystrophie de Schnyder familiale chez une mère (a) et son fils de 18 ans (b).
Transmission autosomique dominante de la mutation du gène DCN au niveau du locus 12q21.33 classant cette dystrophie dans la catégorie 1 [20].
Les opacités cornéennes sont diffuses et bilatérales avant la naissance. Les opacités stromales blanchâtres, en forme de flocons se multiplient avec l’âge, impactant progressivement la vision [21].
Les cellules épithéliales semblent normales et la réflectivité accrue du stroma antérieur empêche de visualiser les autres couches plus profondes. L’évolution est lentement progressive.
Transmission autosomique dominante du gène PIKFYVE au niveau du locus 2q34 (anciennement appelé PIP5K3) classant cette dystrophie dans la catégorie 1.
Le début des signes cliniques est congénital ou néonatal. Cette dystrophie est asymptomatique, parfois associée à une légère photophobie.
Atteinte stromale : présence de petites opacités discoïdes, d’aspect floconneux ou nuageux, translucides et discrètes, plates, de couleur gris-blanc et dispersées à travers tout le stroma dans le 1/3 central de la cornée. Ces mouchetures (flecks) peuvent s’étendre au limbe et sont mieux détectées en rétro-illumination.
L’épithélium, la couche de Bowman, la membrane de Descemet et l’endothélium ne sont pas atteints.
On observe une accumulation de matériaux hyperréflectifs de 2 à 18 mm dans le stroma [22].
Cette dystrophie n’évolue pas avec le temps.
La transmission est autosomique dominante au niveau du locus 12q21.33 par suppression de gènes kératocane (KERA), lumican (LUM), décorine (DCN) et epiphycan (EPYC), classant cette dystrophie dans la catégorie 1 [23].
Le début est néonatal dès 16 semaines ou congénital. Elle est le plus souvent diagnostiquée dans les dix premières années de vie.
Atteinte stromale : cette dystrophie se manifeste par des opacités diffuses, de couleur gris-blanc, en forme de feuille qui peuvent impliquer toutes les couches du stroma mais le plus souvent le stroma postérieur. Les lésions peuvent s’étendre jusqu’au limbe ou être isolées en périphérie, dans les formes les moins sévères.
Les autres anomalies associées sont une diminution de l’épaisseur cornéenne (< 380 μm), une kératométrie plate (< 41 D) et une hypermétropie associée.
Des anomalies iriennes peuvent être présentes : adhérences iridocornéennes, correctopie et polycorie, en particulier chez les patients présentant la forme étendue.
On observe la présence de plis au niveau de la Descemet et d’une couche hyperréflective dans le stroma postérieur.
Lentement progressive, l’acuité visuelle n’est que très légèrement affectée.
Le mode de transmission n’est pas connu, classant cette dystrophie dans la catégorie 4.
Le début des signes cliniques est pendant la première décennie (le patient le plus jeune touché était âgé de 8 ans).
Atteinte stromale : la découverte d’opacités stromales polygonales ou arrondies, centrales, nuageuses, asymptomatiques et entourées de tissu clair est souvent fortuite.
Non progressive. Les modifications de la cornée sont très similaires à l’aspect postérieur « en peau de crocodile » de Vogt, maladie cornéenne dégénérative.
Deux types distincts de CHED ont été décrits : CHED1 (forme lentement progressive, gène situé sur le locus PPCD1 du chromosome 20 comme les PPCD, classifiée comme une mutation autosomique dominante) et CHED2 (forme autosomique récessive du gène SLC4A11, classant cette dystrophie en catégorie 1 si la mutation est retrouvée chez les enfants ou en catégorie 3 pour les enfants sans mutations SLC4A11 [24]).
Cependant, la nouvelle classification IC3D-2, par manque de preuve démontrant l’existence d’une transmission autosomique dominante pour la dystrophie congénitale endothéliale héréditaire, a exclu la CHED autosomique dominante, anciennement connue sous le nom CHED1. Ainsi, la forme autosomique récessive de CHED, anciennement connue sous le nom CHED2, a été renommée CHED.
Le début des signes est congénital ou néonatal. Les nouveau-nés peuvent présenter un nystagmus et, plus tard, les enfants pourront présenter une baisse précoce de l’acuité visuelle.
Atteinte endothéliale et de la membrane de Descemet : oedème de cornée diffus bilatéral (stroma épaissi), souvent asymétrique, avec des plis de la membrane de Descemet et une opacification cornéenne diffuse ; la cornée prenant une coloration bleu-gris (aspect laiteux), pouvant aller jusqu’à un aspect de verre dépoli, qui est souvent présent dès la naissance. Les lésions cornéennes peuvent évoluer vers une opacification totale de la cornée (fig. 9-5).
Histologiquement, il existe un oedème de cornée associé à une raréfaction des cellules endothéliales, parfois empilées, d’aspect multinucléé avec une fine membrane de Descemet.
Le tableau clinique peut se compliquer d’une amyloïdose secondaire sous la forme de dépôts sous-épithéliaux muriformes évoquant le diagnostic de dystrophie gélatineuse en gouttes.
Vision floue dès la naissance avec nystagmus, pouvant nécessiter une prise en charge chirurgicale, comme récemment décrit des greffes endothéliales (Descemet stripping automated endothelial keratoplasty [DSAEK]).
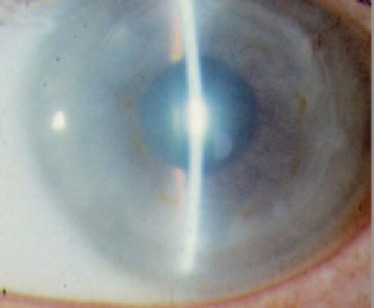
Fig. 9-5 Dystrophie congénitale endothéliale héréditaire.
La dystrophie endothéliale entraîne très rapidement un oedème cornéen, qui empêche progressivement l’analyse du segment antérieur.
(Remerciements au Pr C. Burillon.)
La transmission est autosomique dominante, plusieurs gènes sont impliqués en fonction des différentes formes. Pour la PPCD1, le gène est non connu mais implique le locus 20p11.2-q11.2 (catégorie 2). Pour la PPCD2, les gènes impliqués sont COL8A2 (1p34.2-p32.3) et VSX1 (20p11.21), et pour PPCD3, le gène impliqué est ZEB1 (10p11.22). Ces trois gènes classent cette dystrophie en catégorie 1.
Plusieurs cas précoces voire congénitaux ont été décrits, mais comme cette pathologie est longtemps asymptomatique, elle est diagnostiquée souvent à partir de la troisième décennie chez 50 % des patients.
Le principal diagnostic différentiel est le glaucome congénital, mais il n’existe ni mégalocornée ni excavation papillaire. Les signes cliniques observés sont souvent bilatéraux et asymétriques.
Atteinte endothéliale et de la membrane de Descemet : présence d’opacités grises et géographiques au niveau de la membrane de Descemet et de l’endothélium, qui entourent des lésions vésiculaires simples ou regroupées. On note aussi la présence de bandes endothéliales gris-blanc de forme parallèle (pistes de chemin de fer) qui peuvent s’étendre à travers la totalité de la cornée [[25]] (fig. 9-6).
Il peut exister des adhérences iridocornéennes périphériques dans environ 25 % des cas et une élévation de la pression intraoculaire dans environ 15 % des cas.
Les lésions vésiculaires au niveau de l’endothélium correspondent à des zones sombres et arrondies donnant une apparence en forme de beignet.
Les lésions endothéliales sont en général stables. Une progression lente des vésicules polymorphes au cours des années provoquant parfois une décompensation endothéliale est possible, ce qui nécessite une greffe de cornée chez environ 20 à 25 % des patients touchés.
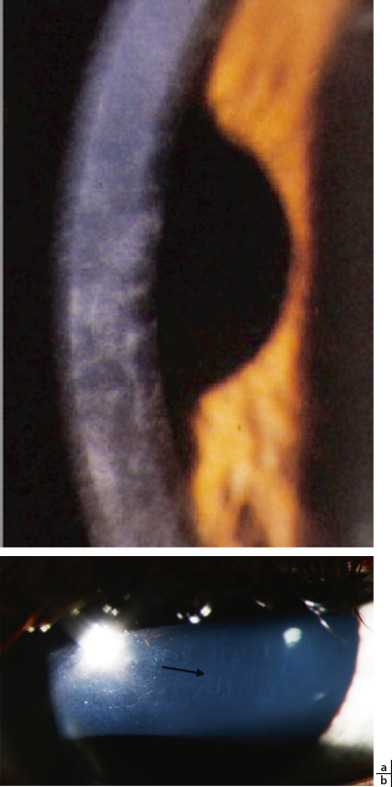
Fig. 9-6 Dystrophie postérieure polymorphe.
a. Atteinte uniquement de l’endothéliodescemet avec peu de signes fonctionnels chez l’enfant. b. À la lampe à fente, aspect de lésions translucides peignées (flèche) avec diminution, voire disparition des cellules endothéliales à ce niveau. (Remerciements au Pr C. Burillon.)
La XECD a été localisée sur le bras long du chromosome X (Xq25) mais le gène causal n’a pas été identifié, classant cette dystrophie dans la catégorie 2.
Le début des signes cliniques est congénital. Les garçons ont souvent une vision floue avec un possible nystagmus, tandis que les filles présentent des formes asymptomatiques.
Atteinte endothéliale : il existe, chez les garçons, une opacification congénitale de la cornée allant d’une opacification diffuse à un aspect en verre dépoli, d’apparence laiteuse, avec présence de « cratères » endothéliaux.
Dans les cas avancés, on observe une kératopathie en bandelette sous-épithéliale, associée à des changements endothéliaux qui ont l’aspect de cratères de lune [26].
Discontinuités focales et changements dégénératifs dans les cellules endothéliales cornéennes sont significatifs de l’apparition de cratères observée sur le plan clinique. La membrane de Descemet est irrégulièrement épaissie avec des petits trous et excavations. L’épithélium cornéen et la couche de Bowman peuvent être irrégulièrement amincis.
Peu progressive chez le garçon. Les filles sont asymptomatiques mais présentent des anomalies endothéliales à type de cratère. Une kératoplastie pénétrante peut être indiquée chez les patients de sexe masculin lorsque l’opacification de la cornée altère significativement la vision.
Les diagnostics différentiels principaux des dystrophies cornéennes correspondent aux opacités congénitales et/ou néonatales cornéennes [13, 27] : pathologies de surcharge (cystinose, tyrosémie type 2, maladies de surcharge lysosomales systémiques telles mucopolysaccharidoses, lipidoses, mucolipidoses, etc.) ; étiologies acquises traumatiques (ruptures de la Descemet verticales et secondaires au forceps) ; kératites infectieuses ; dysgénésies du segment antérieur (par exemple une anomalie de Peters ou un syndrome d’Axenfeld-Rieger compliqué d’un glaucome secondaire) (voir tableau 11-1).
Le glaucome congénital, dont un des signes cliniques est l’oedème cornéen, est également un des diagnostics différentiels principaux des dystrophies de cornée endothéliale (PPCD). Il sera donc toujours à redouter à cause de sa sévérité.
Les indications des kératoplasties pédiatriques incluent :
- les opacités cornéennes bilatérales ;
- les oedèmes cornéens chroniques ;
- les cicatrices cornéennes.
De rares cas de dystrophies cornéennes pédiatriques nécessitent une intervention chirurgicale précoce ; en effet, la baisse d’acuité visuelle est souvent tardive (dystrophies épithéliales stromales TGFB1 ; RBCD ; dystrophies granulaire, grillagée ou maculaire stromale). L’intervention sera alors réalisée à l’âge adulte.
Cependant, certaines formes de dystrophies endothéliales (CHED et dystrophie postérieure polymorphe) peuvent nécessiter une intervention précoce. Les défis des kératoplasties en pédiatrie sont multiples [28] :
- en préopératoire en raison de l’évaluation délicate d’une acuité visuelle chez des enfants en âge préverbal ;
- en peropératoire, les contraintes chirurgicales sont dues :
- à un segment antérieur étroit ;
- aux diamètres cornéens réduits (10 mm avant l’âge de 1 an) ;
- au statut cristallinien ;
- à l’élasticité sclérale qui peut être responsable d’un déplacement antérieur du cristallin ou du diaphragme de l’iris, en raison de la diminution de la pression intra-oculaire peropératoire ;
- à la spécificité des techniques chirurgicales.
- en postopératoire, les difficultés sont dues :
- au taux de rejet élevé et précoce ;
- à la moindre sensibilité au traitement immunosuppresseur [29] ;
- au considérable retard diagnostique de toute complication de greffe, lié à la difficulté des enfants à communiquer la douleur ou à une éventuelle baisse d’acuité visuelle ;
- à la gestion de l’amblyopie : elle sera indispensable car même en présence d’un succès anatomique, elle limitera la récupération fonctionnelle.
Les contre-indications des greffes de cornée chez les enfants sont :
- une amblyopie sévère unilatérale avec un oeil controlatéral sain ;
- un glaucome non contrôlé (risquant un taux d’échec de la kératoplastie transfixiante de 30 % à 1 an [28]) ;
- une kératite infectieuse ou une inflammation active ;
- une mauvaise ou une absence de coopération des parents et de l’entourage.
Grâce aux progrès des techniques chirurgicales et la simplification des soins postopératoires, on observe une augmentation du nombre de kératoplasties transfixiantes réalisées chez les enfants. Le taux de survie du greffon est variable en fonction de l’atteinte et de la technique [30] : les kératoplasties transfixiantes pédiatriques concernent les dystrophies CHED dans 21 % des cas [28] et le taux de survie du greffon à 1 an est estimé à 54 % avec un âge médian (2,1 ans) au moment de la chirurgie. Aucune différence significative n’a été retrouvée entre les groupes d’enfants greffés avant 6 mois, entre 6 mois et 5 ans ou après 5 ans [28]. L’âge de réalisation de la chirurgie est donc à adapter au cas par cas, en tenant compte des caractéristiques de l’enfant et de l’entourage, afin d’espérer une détection précoce des complications postopératoires et une gestion réussie de l’amblyopie [31]. Une autre étude a observé le taux de rejet endothélial après kératoplastie transfixiante pédiatrique de 1998 à 2008 chez 35 enfants présentant une dystrophie endothéliale de type CHED. Chez les enfants de moins de 12 ans, le taux de survie du greffon à 5 ans était de 55 % , 39 % avaient présenté au moins un épisode de rejet endothélial [32].
Les kératoplasties endothéliales seraient une alternative intéressante à la kératoplastie transfixiante (KT) pour les dystrophies endothéliales, car elles entraînent moins d’astigmatisme postopératoire et donc une récupération plus précoce de l’acuité visuelle (6 à 12 semaines après DSAEK versus 6 à 12 mois après KT [31]) et un taux de rejet endothélial réduit.
Une série de DSAEK a été rapportée chez 8 enfants [33], avec un âge moyen de 9 ans au moment de la chirurgie et un suivi moyen de 15,9 mois. Parmi les 3 enfants en bas âge, 2 avaient une fixation en préopératoire, alors que tous (6 yeux) étaient capables de fixer une semaine après la chirurgie. Chez les plus âgés, la meilleure acuité visuelle corrigée était égale ou inférieure à 20/200 en préopératoire, alors qu’en postopératoire, 8 yeux ont eu une meilleure acuité visuelle corrigée, supérieure ou égale à 20/40. Les kératoplasties endothéliales permettraient de restaurer rapidement la transparence cornéenne, en minimisant les complications postopératoires. Ces procédures peuvent être réalisées chez les très jeunes patients. Un cas de DSAEK bilatérale a été rapporté chez un nouveau-né de 8 mois présentant un oedème de cornée congénital secondaire à une dystrophie postérieure polymorphe [34]. Les principales complications rapportées sont la perte de cellules endothéliales du donneur (jusqu’à 50 % ) ou la dislocation du greffon (1 à 2 % rapportées selon les études [35]) qui peuvent conduire à une nouvelle greffe. Les greffes endothéliales semblent être un traitement de choix pour les dystrophies endothéliales chez les enfants qui restent des pathologies rares, pour lesquelles peu de cas ont été décrits à ce jour [36, 37].
[1] You JY, Botelho PJ. Corneal in vivo confocal microscopy : clinical applications. R I Med J (2013) 2016 ; 99 : 30-3.
[2] Rousseau A, Labbé A, Baudouin C, et al. In vivo confocal microscopy and spectral domain anterior segment OCT in Lisch epithelial corneal dystrophy. J Fr Ophtalmol 2015 ; 38 : e151-3.
[3] Weiss JS, Møller HU, Aldave AJ. IC3D Classification of Corneal Dystrophies – Edition 2. Cornea 2015 ; 34 : 117-59.
[4] Nischal Ken K. Genetics of congenital corneal opacification – Impact on diagnosis and treatment. Cornea 2015 ; 34 : S24-34.
[5] Dollfus H, Pelletier V. Génétique et oeil. Encycl Med Chir (Elsevier, Paris). 21-001- A-10. 2016.
[6] Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies Nat Genet 1997 ; 15 : 247-51.
[7] Klinworth GK. Corneal dystrophies. Orphanet J Rare Dis 2009 ; 4 : 7.
[8] Allen EH, Atkinson SD, Liao H, et al. Allele-specific siRNA silencing for the common keratin 12 founder mutation in Meesmann epithelial corneal dystrophy. Invest Ophthalmol Vis Sci 2013 ; 54 : 494-502.
[9] Sullivan LS, Baylin EB, Font R, et al. A novel mutation of the Keratin 12 gene responsible for a severe phenotype of Meesmann’s corneal dystrophy. Mol Vis 2007 ; 13 : 975-80.
[10] Tuft S, Bron AJ. Imaging the microstructural abnormalities of Meesmann corneal dystrophy by in vivo confocal microscopy. Cornea 2006 ; 25 : 868-70.
[11] Lisch W, Büttner A, Offner F, et al. Lisch corneal dystrophy is genetically distinct from Meesmann corneal dystrophy and maps to Xp22.3. Am J Ophthalmol 2000 ; 130 : 461-8.
[12] Kurbanyan K, Sejpal KD, Aldave AJ, et al. In vivo confocal microscopic findings in Lisch corneal dystrophy. Cornea 2012 ; 31 : 437-41.
[13] Hammar B, Lagali N, Ek S, et al. Dystrophia Smolandiensis : a novel morphological picture of recurrent corneal erosions. Acta Ophthalmol 2010 ; 88 : 394-400.
[14] Liang Q, Pan Z, Sun X, Baudouin C, et al. Reis-Bücklers corneal dystrophy : a reappraisal using in vivo and ex vivo imaging techniques. Ophthalmic Res 2014 ; 51 : 187-95.
[15] Chen YJ, Chen JT, Lu DW, et al. In vivo corneal confocal microscopic findings and gene analysis of three patients with Thiel-Behnke corneal dystrophy. Br J Ophthalmol 2010 ; 94 : 262-4.
[16] Han KE, Chung WS, Kim T, et al. Changes of clinical manifestation of granular corneal deposits because of recurrent corneal erosion in granular corneal dystrophy types 1 and 2. Cornea 2013 ; 32 : e113-e120.
[17] Klintworth GK, Bao W, Afshari NA, et al. Two mutations in the TGFBI (BIGH3) gene associated with lattice corneal dystrophy in an extensively studied family. Invest Ophthalmol Vis Sci 2004 ; 45 : 1382-8.
[18] Nowinska AK, Wylegala E, Teper S, et al. Phenotype and genotype analysis in patients with macular corneal dystrophy. Br J Ophthalmol 2014 ; 98 : 1514-21.
[19] Weiss JS, Khemichian AJ. Differential diagnosis of Schnyder corneal dystrophy. Dev Ophthalmol 2011 ; 48 : 67-96.
[20] Bredrup C, Stang E, Bruland O, et al. Decorin accumulation contributes to the stromal opacities found in congenital stromal corneal dystrophy. Invest Ophthalmol Vis Sci 2010 ; 51 : 5578-82.
[21] Jing Y, Kumar RP, Zhu L, et al. Novel decorin mutation in a Chinese family with congenital stromal corneal dystrophy. Cornea 2014 ; 33 : 288-93.
[22] Can E, Kan E, Akgün Hİ. Clinical features and in vivo confocal microscopic imaging of fleck corneal dystrophy. Semin Ophthalmol 2013 ; 28 : 239-41.
[23] Aldave AJ, Rosenwasser GO, Yellore VS, et al. Linkage of posterior amorphous corneal dystrophy to chromosome 12q21.33 and exclusion of coding region mutations in KERA, LUM, DCN and EPYC. Invest Ophthalmol Vis Sci 2010 ; 51 : 4006-12.
[24] Jiao X, Sultana A, Garg P, et al. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet 2007 ; 44 : 64-8.
[25] Aldave AJ, Ann LB, Frausto RF, et al. Classification of posterior polymorphous corneal dystrophy as a corneal ectatic disorder following confirmation of associated significant corneal steepening. JAMA Ophthalmol 2013 ; 131 : 1583-90.
[26] Schmid E, Lisch W, Philipp W, et al. A new, X-linked endothelial corneal dystrophy. Am J Ophthalmol 2006 ; 141 : 478-87.
[27] Shigeyasu C, Yamada M, Mizuno Y, et al. Clinical features of anterior segment dysgenesis associated with congenital corneal opacities. Cornea 2012 ; 31 : 293-8.
[28] Huang C, O’Hara M, Mannis MJ. primary pediatric keratoplasty : indications and outcomes. Cornea 2009 ; 28 : 1003-8.
[29] Stulting RD, Sumers KD, Cavanagh HD, et al. Penetrating keratoplasty in children. Ophthalmology 1984 ; 91 : 1222-30.
[30] Al-Ghamdi A, Al-Rajhi A, Wagoner MD. Primary pediatric keratoplasty : indications, graft survival, and visual outcome. J AAPOS 2007 ; 11 : 41-7.
[31] Javadi MA, Baradan-Rafii AR, Zamani M, et al. Penetrating keratoplasty in young children with congenital hereditary endothelial dystrophy. Cornea 2003 ; 22 : 420-3.
[32] Özdemir B, Kubaloglu A, Koytak A, et al. Penetrating keratoplasty in congenital hereditary endothelial dystrophy. Cornea 2012 ; 31 : 359-65.
[33] Busin M, Beltz J, Scorcia V. Descemet-stripping automated endothelial keratoplasty for congenital hereditary endothelial dystrophy. Arch Ophthalmol 2011 ; 129 : 1140-6.
[34] Sella R, Rootman D, Bahar I. Descemet’s stripping automated endothelial keratoplasty for posterior polymorphous corneal dystrophy in an 8-month-old boy. J AAPOS 2013 ; 17 : 94-6.
[35] Kathryn Colby K. Changing times for pediatric keratoplasty. J AAPOS 2008 ; 12 : 223-4.
[36] Jeng BH, Marcotty A, Traboulsi E. Descemet stripping automated en dothelial keratoplasty in a 2-year-old child. J AAPOS 2008 ; 12 : 317-8.
[37] Pineda R II, Jain V, Shome D, et al. Descemet’s stripping endothelial keratoplasty : is it an option for congenital hereditary endothelial dystrophy ? Int Ophthalmol 2010 ; 30 : 307-10.
C. Burillon
Le kératocône (KC) est la plus connue des maladies ectatiques de la cornée et apparaît le plus souvent dans la seconde décennie de la vie ; l’évolution est progressive jusqu’à la troisième décennie, puis se stabilise.
Son nom vient d’une racine grecque : keras (= cornée) et konos (= cône) [1]. C’est donc une dystrophie cornéenne qui entraîne un amincissement progressif de la cornée qui se déforme en un cône bombé en avant. Cette distorsion de la cornée est responsable de l’apparition d’une myopie et d’un astigmatisme irrégulier, dans un premier temps, puis la réduction de l’acuité visuelle peut devenir irréversible en raison de l’apparition de cicatrices cornéennes ou d’oedème.
Il est difficile d’évaluer la prévalence du KC, variant de 8,8 à 54,4 pour 100 000 dans les publications avant 2007 (diagnostic porté essentiellement sur la kératométrie) et de 760 à 3300 pour 100 000 dans celles après 2009, avec utilisation systématique du topographe cornéen pour faire le diagnostic [1]. Cette prévalence est plus basse chez les Caucasiens que les Asiatiques, même si le KC est connu pour toucher toutes les ethnies [2].
Des facteurs à la fois environnementaux et génétiques contribuent à sa pathogénie.
Longtemps considérée comme une affection non inflammatoire, la mise en évidence récente de la surexpression de médiateurs de l’inflammation comme les cytokines et l’interleukine 6 (IL-6), dans les larmes de patients présentant un KC déclaré ou infraclinique, remet en question ce concept et oriente la recherche pathogénique dans ce sens [3, 4].
Les facteurs environnementaux sont bien décrits dans la littérature. Ils devraient permettre la reconnaissance très précoce d’un KC chez l’enfant et être une aide thérapeutique précieuse.
Le frottement oculaire est un facteur retrouvé de façon très élevé chez l’enfant porteur de KC, jusqu’à 91,84 % dans l’étude bordelaise de 2012 [5]. Il peut être à l’origine d’aggravation du KC, allant parfois jusqu’à faciliter l’apparition d’un hydrops cornéen [6] chez des enfants en dessous de 10 ans. Il peut cependant préexister avant le diagnostic de KC. Certains évoquent le rôle direct du frottement oculaire répété dans la genèse du KC [7].
Les microtraumatismes induits par le frottement sur l’épithélium entraînent une augmentation des métalloprotéinases (1 et 13) et l’apparition de facteurs médiateurs de l’inflammation comme l’IL-6 et le tumor necrosis factor α (TNF-α) [8], qui ont une part de responsabilité dans le développement du KC.
L’atopie est retrouvée chez un tiers des patients présentant un KC, s’exprimant sous la forme d’asthme, d’eczéma et d’allergies diverses. Le frottement oculaire lié à une allergie oculaire va s’intriquer dans la pathogénie du KC.
L’exposition solaire avec la lumière ultraviolette entraîne des dommages oxydatifs de la cornée, avec une réduction conséquente du taux d’aldéhyde déshydrogénase et de superoxyde dismutase, enzymes nécessaires pour détruire les radicaux libres. Un amincissement du stroma cornéen avec une perte de kératocytes est retrouvé chez les souris exposées aux UV [9] et l’amincissement stromal caractérise les populations vivant dans les pays exposés au soleil [1], comme Israël, l’Arabie saoudite, le Liban, l’Inde et l’Iran, à la différence des pays nordiques. Cette variation d’épaisseur existe en Europe entre les pays du sud et ceux du nord. Cependant la notion d’exposition aux UV ne suffit pas à expliquer les variations géographiques : en effet, le KC est plus fréquent chez les Perses d’Iran que chez les non-Perses (Arabes, Turcs, Kurdes) vivant en Iran [10], et l’âge d’apparition est plus précoce chez les Asiatiques que les Caucasiens.
Les facteurs génétiques sont donc bien présents, jouant un rôle important dans la pathogénie du KC. Ils ne semblent pas relever de la mutation d’un seul gène, comme dans les maladies mendéliennes, mais plutôt de la variation de nombreux gènes situés sur des loci différents. La transmission est cependant souvent autosomique dominante à pénétrance variable, parfois autosomique récessive. Une des preuves de l’étiologie génétique est la présence d’autres personnes atteintes dans la famille, soit sous une forme familiale connue, soit sous une forme mineure qu’il va falloir rechercher. Ce diagnostic de KC frustre est facilité par l’examen de la topographie cornéenne qui a changé notre prise en charge depuis son développement. La preuve de l’étiologie génétique est également retenue du fait de l’existence de KC chez deux jumeaux homozygotes, ou hétérozygotes, avec des moments d’apparition et des niveaux de sévérité qui peuvent rester différents [11] et par l’association possible du KC avec d’autres maladies génétiques. Le syndrome de Down est le plus connu avec une fréquence de 5 à 6 % de KC reconnus [12]. Le syndrome de Turner et le syndrome d’hypopigmentation généralisée présentent parfois un KC. L’amaurose congénitale de Leber, le syndrome d’Ehlers-Danlos, l’ostéogenèse imparfaite, le syndrome de Marfan, le prolapsus de la valve mitrale sont en relation avec une maladie des protéines de la matrice extracellulaire et peuvent être associés à un KC, mais également la maladie de Crouzon, le syndrome de Noonan, la syndactylie, le syndrome de Raynaud, et la fièvre méditerranéenne familiale [13].
Toutes ces affections malformatives reconnues comme génétiques devraient faire l’objet d’un examen ophtalmologique complet, le plus tôt possible, à la recherche en particulier de KC.
L’identification des gènes responsables du KC a fait l’objet de nombreuses publications.
La plus récente (2016) et complète provient de l’équipe de Yaron Rabinowitz qui travaille depuis très longtemps sur le kératocône [14]. Grâce à l’utilisation d’outils génétiques complets (études de liaison génétique et d’association génétique par homozygotie par filiation), la recherche d’un marqueur chromosomique associé à un KC, dans des familles atteintes et/ou consanguines, permet de réaliser une cartographie génétique. L’utilisation de puces SNP (single nucleotide polymorphism) donne la localisation du ou des gènes sur le chromosome. Dans le KC, des mutations des gènes suivants mises en évidence par les puces SNP sont impliquées : LOX, CAST, DOCK9, IL1RN, SLC4A11, HGF, RAB3GAP1, TGFB1, ZNF469, ZEB1, VSX1, COL5A1, COL4A3, COL4A4, FNDC3B, FOXO1, MPDZ-NF1B, WNT10A, SOD1, IL1B, IL1A et microRNA MIR184. Cette liste établie par Rabinowitz n’est certainement pas exhaustive.
Ces gènes sont localisés sur des portions de chromosome connues. Nous ne détaillerons que les trois plus consensuels à ce jour. LOX (lysyl oxidase) s’exprime sur la portion 5q23.2 du chromosome 5 et a pour rôle d’initier le cross-linking des fibres de collagène et d’élastine par désamination oxydative catalysante du groupe epsilon- amino dans certains résidus lysine et hydroxylysine. Son absence peut réduire le cross-linking des fibres de collagène cornéen, déséquilibrant la biomécanique du stroma [15]. Le CAST, gène de la calpastatine, se situe sur la région 5q15 et permet l’inhibition de la calpaïne, protéase intracellulaire. Cette calpastatine participe à la rigidité du tissu [16]. Plusieurs isoformes de CAST sont retrouvées dans l’oeil (cornée, cristallin, ptérygion). Le DOCK9 (dedicator of cytokinesis 9) est localisé sur le 13q32.3 et sa mutation est rapportée dans plusieurs familles équatoriennes présentant un KC [17].
L’étude des origines génétiques complexes du KC a montré que les différents troubles du tissu conjonctif peuvent avoir les mêmes déterminants génétiques : le brittle cornea syndrome, qui est une maladie autosomique récessive, généralisée au tissu conjonctif, présente une cornée très fine avec un risque important de rupture ; ce sont des mutations du gène ZNF469 (zinc finger protein 469), situé en 16q24.2, qui induisent cette maladie, avec un risque élevé d’apparition d’un KC [18]. De la même façon, les mutations du gène TGFB1 (kérato-épithéline) sont retrouvées dans certaines dystrophies épithéliales ou stromales cornéennes, dans les amyloses cornéennes primitives ou secondaires, mais également dans certaines populations chinoises [19] et polonaises de KC [20]. Le locus concerné est situé en 5q31.1.
Ainsi, le KC atteint indifféremment les hommes et les femmes, et toutes les ethnies dans le monde. Des facteurs environnementaux et génétiques contribuent à sa pathogénie. Les nouveaux développements des techniques génétiques de « sequencing » sont prometteurs et devraient améliorer nos connaissances sur cette origine du KC, permettant ainsi un dépistage plus précoce et une thérapie plus ciblée dans les années à venir [1].
Le KC de l’enfant n’est jamais diagnostiqué assez tôt. Si 73 % des diagnostics sont faits avant l’âge de 24 ans [12], la plainte de l’enfant est toujours modérée voire inexistante, et l’ophtalmologiste pense rarement à cette pathologie avant la puberté.
Devant l’apparition d’une myopie, et encore plus si elle est associée à un astigmatisme myopique, une topographie cornéenne devrait toujours être réalisée chez l’enfant, même au plus jeune âge.
Les signes cliniques sont semblables à l’affection qui atteint l’adulte. Il ne sera souligné ici que ce qui est très spécifique de l’enfant.
La baisse d’acuité visuelle est souvent asymétrique, voire unilatérale stricte dans les rares cas, chez l’enfant, de KC unilatéral. La vision de près est préservée pendant longtemps, grâce à la myopie et l’astigmatisme le plus souvent inverse qui résultent de la déformation kératoconique.
Une photophobie, l’impression d’un brouillard visuel variable mais progressif, une irritation oculaire, qui va entraîner un frottement chronique, sont des signes fonctionnels plus difficiles à mettre en évidence chez l’enfant mais tout à fait suspects d’une atteinte cornéenne.
À l’examen, le signe de Munson (déformation angulaire de la paupière inférieure lors de l’abaissement du globe) est souvent retrouvé, en particulier dans des cas évolués d’enfants avec retard psychomoteur. Un strabisme peut apparaître lorsqu’un oeil est très atteint et que la baisse de vision est ancienne. Les autres signes (signe Rizzuti, anneau de Fleischer) ne sont plus vraiment recherchés et l’examen biomicroscopique est souvent peu informatif, sauf en cas de KC évolué (fig. 9-7). L’examen à la lampe à fente peut mettre en évidence l’amincissement cornéen au sommet du cône, une visibilité anormale des nerfs cornéens, des lignes cicatricielles superficielles (aspect réticulaire au niveau de la membrane de Bowman, probables ruptures de cette membrane) ou profondes, volontiers au sommet de l’ectasie (fig. 9-8), à distinguer des stries de Vogt, lignes de contraintes prédescemétiques, alignées le long du méridien à plus grande courbure.
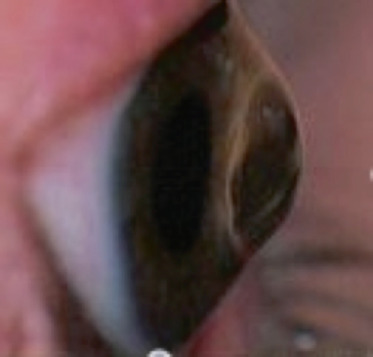
Fig. 9-7 Kératocône évolué bien visible de profil.
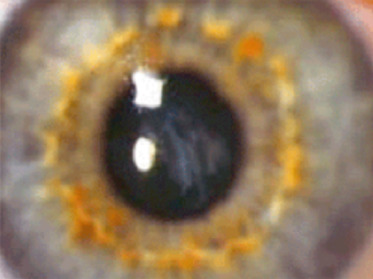
Fig. 9-8 Cicatrices sommitales cornéennes de kératocône.
L’examen à la lampe à fente permettra d’éliminer une autre pathologie oculaire, comme une cataracte ou une luxation du cristallin, et de faire le diagnostic différentiel avec une mégalocornée ou un kératoglobe. Les opacités des kératoconjonctivites de la petite enfance et les séquelles de trachome peuvent déformer la cornée, lui donnant un aspect pseudo-kératoconique qu’il faudra distinguer d’un vrai KC. Tous les facteurs favorisants l’apparition du KC seront recherchés (atopie en particulier).
L’évolution spontanée se fait vers la déformation de plus en plus importante de la cornée, avec une kératométrie qui peut dépasser 70 D et une meilleure acuité visuelle corrigée qui atteint juste 1/10. À ces stades évolués, l’apparition possible d’un KC aigu ou hydrops est la conséquence de la rupture de la membrane de Descemet, en raison de la cambrure trop importante de la cornée. L’humeur aqueuse pénètre alors dans le stroma cornéen, entraînant un oedème stromal brutal et l’apparition d’une opacité cornéenne. Les signes fonctionnels sont bruyants, avec une photophobie douloureuse, un oeil larmoyant et une conjonctive hyperhémiée. L’évolution se fait vers la résorption de l’oedème grâce à la migration des cellules endothéliales en regard de la brèche descemétique (fig. 9-9). Un léger aplatissement cornéen séquellaire peut autoriser une amélioration de la vision si la cicatrice fibreuse n’est pas centrale. Dans le cas contraire, la baisse de vision est inéluctable.
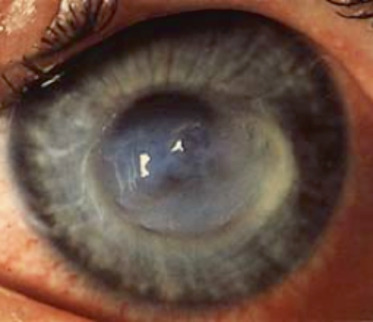
Fig. 9-9 Kératocône aigu ou hydrops.
Parfois le diagnostic d’hydrops n’est pas fait immédiatement devant des signes évoquant plus une kératite infectieuse, comme dans ce cas d’un enfant de 10 ans, dont la maladie kératoconique a évolué rapidement et dont le diagnostic ne sera porté que devant l’échec thérapeutique des antibiotiques [21].
Les frottements répétés de l’oeil en raison d’un terrain atopique [22] ou d’une kératoconjonctivite vernale [6] peuvent entraîner un hydrops bilatéral chez des enfants en dessous de 10 ans. Dans ces deux présentations de cas que nous citons, la pathologie oculaire n’avait pas été suivie et le diagnostic de KC n’avait pas été fait auparavant. Les signes vidéotopographiques ont confirmé une ectasie localisée avec un amincissement considérable de la cornée. Cependant, ne devrait-on pas alors parler de KC secondaire ? Ou mieux, de déformation kératoconique secondaire à une inflammation chronique des yeux de ces petits enfants ?
Au terme de l’examen clinique, lorsque l’ophtalmoscope de Javal était utilisé, la classification d’Amsler permettait de déterminer le degré de gravité, du premier au quatrième, l’amincissement cornéen étant majeur dans le 4e degré, avec un astigmatisme non mesurable car irrégulier et des opacités cornéennes.
Actuellement, l’examen de choix est la vidéotopographie cornéenne d’élévation (Pentacam® et Orbscan® sont les plus utilisés en France), qui permet de déterminer la kératométrie de toute la surface cornéenne, celle de la face postérieure et la pachymétrie de toute la cornée. Elle confirme le diagnostic et l’évoque dans les cas frustres grâce aux différents indices kératocôniques proposés par les topographes cornéens. Elle permet aussi de distinguer une forme clinique particulière du KC qui est la dégénérescence pellucide marginale : l’amincissement cornéen intéresse une bande étroite périphérique, séparée du limbe d’environ 1 mm, généralement dans la partie inférieure de la cornée. L’épaisseur de la cornée centrale est normale. Cette forme est rare chez l’enfant.
La vidéotopographie cornéenne est un examen indispensable qui sera répété tous les 3 mois au début afin de juger de l’évolutivité du KC. Il ne serait pas acceptable de ne pas réaliser cet examen chez un enfant suspect d’avoir un KC.
Avec les valeurs kératométriques et pachymétriques, la classification de Krumeich est alors utilisée pour classer le KC (fig. 9-10 et tableau 9-7).
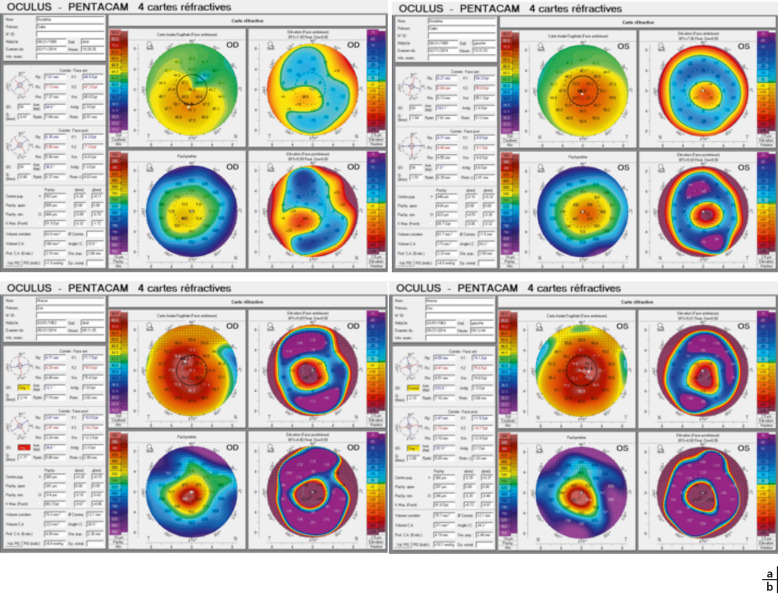
Fig. 9-10 Kératocônes : stades II (a) et IV (b) de Krumeich.
Chez l’enfant, le KC est toujours diagnostiqué à un stade plus sévère que l’adulte : 27,8 % de stade IV chez l’enfant de moins de 15 ans versus 7,8 % au-delà de 27 ans, dans l’étude de Colin en 2012 [23]. Toutes les valeurs kératométriques sont plus élevées que chez l’adulte, au moment du diagnostic. Cela est en relation avec l’absence de plainte de l’enfant qui ne sait pas exprimer qu’il a une baisse d’acuité visuelle et qui va utiliser des attitudes compensatoires pour voir mieux, comme le clignement palpébral créant un trou sténopéïque. Lorsqu’il progresse, le KC évolue beaucoup plus rapidement chez l’enfant, de la même façon qu’il évolue plus vite chez les jeunes adultes (âge moyen 22,2 ans) que chez les plus vieux (24,7 ans), dans une étude coréenne récente [24]. Ainsi la détection doit être la plus précoce possible, avec la réalisation d’une topographie cornéenne très tôt dans le suivi d’un trouble réfractif chez l’enfant. De la même façon, le suivi doit être rapproché, tous les 3 mois, tant qu’on n’a pas fait la preuve de la stabilisation de la maladie.
Du fait de la déformation rapide de la cornée chez l’enfant, les lésions anatomopathologiques, mises en évidence sur une pièce anatomique lors de greffe de cornée, sont d’emblée caractéristiques du KC. L’amincissement cornéen est maximal dans la zone centrale du KC, en rapport avec une réduction du nombre de lamelles de collagène, qui perdent leur parallélisme. L’épithélium est plus ou moins atrophique avec réduction du nombre de couches et la membrane basale présente des lésions de rupture comme la membrane de Bowman, avec épaississement et aspect fibrillaire sur ces zones, signant la présence d’un type de collagène différent, cicatriciel souvent. L’endothélium reste intact très longtemps, jusqu’à l’apparition de polymégathisme, après hydrops ou après le port prolongé de lentilles de contact. La membrane de Descemet est normale jusqu’au stade III inclus, puis présente des encoches ou ruptures lors de l’apparition du KC aigu [12].
Aujourd’hui, la microscopie confocale permet de visualiser des éléments anatomiques et cellulaires in vivo : la densité stromale des kératocytes est abaissée dans le KC évident ou fruste, et le diamètre des fibres nerveuses est augmenté par comparaison avec une population saine de contrôle [25]. La systématisation de cet examen pourrait permettre de conforter le diagnostic de KC dans des stades très initiaux de la maladie, avec une topographie normale.
L’adaptation par lentille de contact est la règle, dès que le diagnostic de KC est fait, quel que soit l’âge. Plus l’enfant est jeune, plus l’adaptation sera facile et rapide. L’adaptation doit être bilatérale même si l’atteinte est principalement unilatérale, car lorsqu’il sera devenu adulte, et quels que soient son évolution et les traitements subis, la correction par lentilles lui permettra d’obtenir une acuité visuelle corrigée optimale. Une paire de lunettes sera également prescrite mais l’enfant se rendra rapidement compte de la médiocrité de cette correction et préférera les lentilles.
Les différentes solutions pour adapter au mieux un enfant porteur de KC doivent associer une sécurité maximale et un confort physique et visuel, respectueux de la physiologie oculaire [26]. Des lentilles rigides spéciales KC sont prescrites en premier, puis des essais en piggy-back avec une lentille souple en silico-hydrogel associée à la lentille rigide sont proposés en cas d’inconfort. Depuis quelques années, les lentilles sclérocornéennes, sans contact cornéen et à appui scléral, permettent de mieux corriger le défaut optique avec une oxygénation maximale cornéolimbique et sans les effets secondaires des appuis cornéens non souhaités. Des lubrifiants sans conservateurs peuvent être instillés, surtout s’il existe un terrain atopique sous-jacent.
Le cross-linking cornéen (CXL) est indiqué dès qu’il y a évolution du KC. Le CXL utilise des rayons UV et de la riboflavine, vitamine B2 photosensitive. La réaction photochimique entre les deux facilite le développement de liaisons chimiques entre les fibres de collagène stromal, rigidifiant ainsi la cornée. Le KC stoppe alors son évolution. La riboflavine doit pénétrer le plus profondément dans le stroma cornéen afin que la formation des liaisons interfibres ne soit pas située uniquement dans le stroma antérieur. Dans ce cas, il y aurait échappement au traitement après quelques mois ou années et obligation de recommencer.
Chez l’enfant, cette indication doit être rapidement posée, 3 mois après le diagnostic de KC, si la kératométrie se modifie, même un peu, quel que soit son âge. Aucune norme n’est actuellement établie, mais la tendance est de faire systématiquement un cross-linking chez un enfant ou adolescent de moins de 18 ans en raison de l’évolutivité certaine du KC en dessous de cet âge.
Les publications du CXL chez l’enfant sont plus rares que chez l’adulte [27], mais elles démontrent toutes l’efficacité de cette technique, à la fois sur la stabilisation topographique et l’amélioration sensible de l’acuité visuelle [28, 29]. Le protocole de Dresden (epithelium-off, 30 minutes avec UV 3 MW/cm3) qui commence par l’ablation de l’épithélium cornéen avant l’instillation de la riboflavine est certainement, à ce jour, celui qui donne les meilleurs résultats en raison d’une très bonne pénétration de la riboflavine dans le stroma. Le CXL transépithélial ne semble pas être une technique très performante [30], alors que le CXL accéléré, avec une dose d’UV plus forte sur un temps de 4 minutes seulement, donnerait les mêmes résultats que le CXL classique [31].
Plus récemment, le CXL par iontophorèse, permettant la pénétration de la riboflavine dans la cornée sans enlever l’épithélium, a été réalisé chez l’enfant avec des résultats publiés satisfaisants mais qui ne dépassent pas 24 mois encore [32].
La complication la plus redoutable après CXL est la kératite infectieuse, justifiant la mise sous antibiotique local large spectre jusqu’à cicatrisation totale de l’épithélium.
Bien évidemment, chez l’enfant, l’anesthésie générale est utilisée le plus souvent. Le plus petit enfant ainsi traité dans la littérature est une petite fille atteinte du syndrome de Down, qui a subi un CXL à l’âge de 4 ans pour un oeil puis 5 ans pour l’autre, avec un résultat excellent sur un suivi de 3 ans [33]. Dès que l’enfant est en âge de comprendre et se tenir tranquille, l’anesthésie locale est privilégiée, malgré un temps de traitement qui peut aller jusqu’à 30 minutes.
La greffe de cornée sera proposée lorsqu’il y aura des opacités stromales irréversibles, empêchant toute récupération visuelle, même avec des lentilles. Parmi 65 greffes réalisées chez l’enfant en Nouvelle-Zélande, entre 1991 et 2003, Patel retrouve 67,2 % de KC, 15,5 % de pathologies congénitales et 10,3 % de suites traumatiques. Parmi les 39 yeux greffés porteurs d’un KC, 5 concernaient des enfants entre 5 et 9 ans, 34 entre 10 et 14 ans. Les résultats des kératoplasties transfixiantes étaient satisfaisants, avec plus de 60 % d’acuité visuelle supérieure à 3/10, et 5 rejets seulement (12 % ). Il est bien évident qu’avec la réalisation précoce de CXL, ce nombre de kératoplasties devrait diminuer considérablement [34].
À ce jour, la meilleure indication de greffe de cornée dans le KC est la kératoplastoplastie lamellaire antérieure profonde (KLAP) qui conserve l’endothélio-descemet du petit patient, diminuant ainsi le risque de rejet [35]. Cette technique n’est pas possible chirurgicalement s’il y a eu rupture endothélio-descemétique (hydrops), et ne donne pas de bons résultats visuels si la kératométrie est trop élevée (> 60 D) car il y aura alors la création de plis dans l’interface greffon et Descemet, responsables d’aberrations optiques importantes. La réalisation d’un KLAP permet une récupération visuelle plus rapide, avec une ablation sélective des points séparés, et diminue le risque d’ouverture du globe lors d’un traumatisme oculaire, risque qui n’est pas rare chez l’enfant et l’adolescent. Le changement de greffon s’avère également moins dangereux, puisqu’il n’y aura pas d’inflammation de la chambre antérieure. Il n’y a cependant aucune publication sur ce sujet actuellement. La greffe transfixiante reste encore très utilisée dans les KC stade IV diagnostiqués trop tardivement, avec cicatrices profondes de rupture endothélio-descemétiques.
Les segments d’anneaux intracornéens qui aplatissent la cornée pour autoriser une adaptation en lentilles de contact plus facile n’ont pas encore leur place dans la prise en charge du KC de l’enfant. En effet, soit le diagnostic du KC est précoce et le CXL permet une stabilisation réelle de la maladie jusqu’à l’âge adulte, soit le diagnostic est trop tardif et oblige à la réalisation d’une greffe de cornée. Cependant, dans le cas d’un KC de stade II ou III, avec un CXL fait, l’acuité visuelle doit pouvoir être correctement corrigée avec lunettes ou plus souvent lentilles. Si cette acuité visuelle ne devait pas s’améliorer avec correction optique, l’indication de pose d’anneaux intracornéens trouverait sa place, comme chez l’adulte [27].
Dans tous les cas, la prise en charge du KC de l’enfant doit être la plus précoce possible et l’adaptation en lentilles de contact s’impose, suivie de la réalisation d’un CXL dès qu’on a la preuve de la maladie et de son évolution.
[1] Gordon-Shaag A, Millodot M, Shneor E, Liu Y. The genetic and environmental factors for keratoconus. Biomed Res Int 2015 ; 2015 : 795738.
[2] Nowak DM, Gajecka M. The genetics of keratoconus. MEAJO 2011 ; 18 : 2-6.
[3] Jun AS, Cope L, Speck C, et al. Subnormal cytokine profile in the tear fluid of keratoconus patients. PLoS ONE 2011 ; 6 : el6437.
[4] Davidson AE, Hayes S, Hardcastle AJ, Tuft J. The pathogenesis of keratoconus. Eye 2014 ; 28 : 189-95.
[5] Léoni-Mesplié S, Mortemousque B, Mesplié N, et al. Epidemiological aspects of keratoconus in children. J Fr Ophtalmol 2012 ; 35 : 776-85.
[6] Panahi-Bazaz MR, Sharifipour F, Moghaddasi A. Bilateral keratoconus and corneal hydrops associated with eye rubbing in a 7-year-old girl. J Ophthalmic Vis Res 2014 ; 9 : 101-5.
[7] Carlson AN. Expanding our understanding of eye rubbing and keratoconus. Cornea 2010 ; 29 : 245.
[8] Balasubramanian SA, Pye DC, Willcox MDP. Effects of eye rubbing on the levels of protease, protease activity and cytokines in tears : relevance in keratoconus. Clinical and Experimental Optometry 2013 ; 96 : 214-8.
[9] Newkirk KM, Chandler HL, Parent AE et al. Ultraviolet radiation-induced corneal degeneration in 129 mice. Toxicologic Pathology 2007 ; 35 : 819-26.
[10] Hashemi H, Khabazkhoob M, Fotouhi A. Topographic keratoconus is not rare in an Iranian population : the Tehran eye study. Ophthalmic Epidemiology 2013 ; 20 : 385-91.
[11] Tuft SJ, Hassan H, George S, et al. Keratoconus in 18 pairs of twins. Acta Ophtalmologica 2012 ; 90 : 482-6.
[12] Arné JL. Kératocône. Encycl Méd Chir (Elsevier, Paris). Ophtalmologie, 21-200-D- 40. 2005.
[13] Kosker M, Arslan N, Alp MY, et al. Association between keratoconus and familial mediterranean fever in Turkey. Cornea 2016 ; 35 : 77-80.
[14] Bykhovskaya Y, Margines B, Rabinowitz Y. Genetics in keratoconus : where are we ? Eye and Vision 2016 ; 3 : 16-26.
[15] Bykhovskaya Y, Li X, EpifantsevaI T et al. Variation in the lysyl oxidase (LOX) gene is associated with keratoconus in family-based and case-control studies. Invest Ophthalmol Vis Sci 2012 ; 53 : 4152-7.
[16] Li X, Bykhovskaya Y, Tang YG et al. An association between the calpastatin (CAST) gene and keratoconus. Cornea 2013 ; 32 : 696-701.
[17] Gajecka M, Radhakrishna U, Winters D, et al. Localization of a gene for keratoconus to a 5.6-Mb interval on 13q32. Invest Ophthalmol Vis Sci 2009 ; 50 : 1531-9.
[18] Davidson AE, Borasio E, Liskova P, et al. Brittle cornea syndrome ZNF469 mutation carrier phenotype and segregation analysis of rare ZNF469 variants in familial keratoconus. Invest Ophthalmol Vis Sci 2015 ; 56 : 578-86.
[19] Guan T, Liu C, Ma Z, Ding S. the point mutation and polymorphism in keratoconus candidate gene TGFβI in chinese population. Gene 2012 ; 503 : 137-9.
[20] Karolak JA, Polakowski P, Szaflik J, et al. Molecular screening of keratoconus susceptibility sequence variants in VSX1, TGFβI, DOCK9, STK24, and IPO5 genes in polish patients and novel TGFβI variant identification. Ophthalmic Genet 2016 ; 37 : 37-43.
[21] Slim EA, Jarade EF, Charanek BM, et al. Acute corneal hydrops mimicking infectious keratitis as initial presentation of keratoconus in a 10-year-old child. Case Rep Ophthalmol Med 2015 ; 2015 : 308348.
[22] Downie LE. The necessity for ocular assessment in atopic children : bilateral corneal hydrops in a 8 year old. Pediatrics 2014 ; 134 : 596-601.
[23] Léoni-Mesplié S, Mortemousque B, Touboul D, et al. Scalability and severity of keratoconus in children. Am J Ophthalmol 2012 ; 154 : 56-62.
[24] Ahn SJ, Kim MK, Wee WR. Topographic progression of keratoconus in the Korean population. Korean J Ophthalmol 2013 ; 27 : 162-6.
[25] Ozgurhan EB, Kara N, Yildirim A, et al. Evaluation of corneal microstructure in keratoconus : a confocal microscopy study. Am J Ophtalmol 2013 ; 156 : 885-93.
[26] Mallet F, et al. Les lentilles de contact. Rapport de la Société française d’ophtalmologie. Issy-les-Moulineaux : Masson ; 2009.
[27] Kankariya VP, Kymionis GD, Diakonis VF, Yoo SH. Mamagement of pediatric keratoconus- Evolving role of corneal collagen cross-linking : an update. Indian J Ophthalmol 2013 ; 61 : 435-40.
[28] McAnena L, O’Keefe M. Corneal collagen cross-linking in children with keratoconus. J AAPOS 2015 ; 19 : 228-32.
[29] Chatzis N, Hafezi F. progression of keratoconus and efficacity of pediatric (corrected) corneal collagen cross-linking in children and adolescents. J Refract Surg 2012 ; 28 : 753-8.
[30] Buzzonetti L, Petrocelli G. transepithelial corneal cross-linking in pediatric patients : early results. J Refract Surg 2012 ; 28 : 763-7.
[31] Ozgurhan EB, Kara N, Cankaya KI, et al. Accelerated corneal cross-linking in pediatric patients with keratoconus : 24-month outcomes. J Refract Surg 2014 ; 30 : 843-9.
[32] Buzzonetti L, Petrocelli G, Valente P, et al. Iontophoretic transepithelial corneal crosslinking to halt keratoconus in pediatric cases : 15-month follow-up. Cornea 2015 ; 34 : 512-5.
[33] Sabti S, Tappeiner C, Frueh BH. Corneal cross-linking in a 4-year-old child with keratoconus and down syndrome. Cornea 2015 ; 34 : 1157-60.
[34] Patel HY, Ormonde S, Brookes NH, et al. The indications and outcome of paediatric corneal transplantation in New Zealand : 1991-2003. Br J Ophthalmol 2005 ; 89 : 404-8.
[35] AsharJN, Pahuja S, Ramappa M, et al. Deep anterior lamellar keratoplasty in children. Am J Ophthalmol 2013 ; 155 : 570-4.
C. Guis, L. Hoffart
Dans les pays en voies de développement, 1,5 à 8 000 000 ulcères de cornée sont observés chaque année tous âges confondus [1]. Les études relatives aux kératites microbiennes chez l’enfant sont rares. L’étude d’Ormerod et al. rapporte que la kératite infectieuse chez l’enfant représente 11 % de l’ensemble des cas de kératite microbienne [2]. Elles ont pour principale étiologie les atteintes bactériennes devant les kératites fongiques ou parasitaires. Les kératites infectieuses pédiatriques sont donc peu fréquentes mais présentent un enjeu thérapeutique majeur du fait du risque d’amblyopie secondaire [3].
Les kératites infectieuses pédiatriques diffèrent des formes adultes par une réaction inflammatoire majorée et par un examen clinique pouvant s’avérer difficile selon l’âge du patient. Elles peuvent donc souffrir d’un retard diagnostique et thérapeutique, source éventuelle de séquelles visuelles invalidantes.
L’équipe de Maurin et al. rapporte que l’incidence de la cécité cornéenne chez les enfants vivant dans les pays tropicaux est 20 fois supérieure à celle dans les pays développés [4]. L’incidence des kératites microbiennes est de 113 pour 100 000 habitants en Inde [5] et de 799 pour 100 000 habitants au Népal [6] comparée à 11 pour 100 000 habitants aux États-Unis [7].
Les kératites microbiennes nécessitent l’altération d’un ou de plusieurs des mécanismes impliqués dans la protection contre les agents infectieux du tissu cornéen : intégrité de l’épithélium, normalité du film lacrymal et clignement palpébral efficace. Leur survenue est étroitement liée aux facteurs de risque altérant ces mécanismes de défense. La plupart des agents pathogènes ne pouvant pénétrer un épithélium sain, les kératites infectieuses se produisent le plus fréquemment suite à une atteinte de l’épithélium cornéen.
Les facteurs de risque de kératite infectieuse varient en fonction de la situation géographique et de l’âge. De façon non exhaustive, on peut considérer le port de lentilles de contact, les maladies systémiques dont la carence en vitamine A, les irritations chroniques cornéennes (entropion, kératite sèche, toxicité), les traumatismes oculaires, les chirurgies cornéennes et les traitements par corticostéroïdes topiques comme les principaux facteurs associés. Un facteur de risque est le plus souvent retrouvé (92,6 % des cas) [8] lors d’une kératite infectieuse non virale chez l’enfant avec principalement le traumatisme oculaire dans le groupe des enfants les plus jeunes. La fréquence de ce facteur de risque est variable selon la zone géographique avec par exemple une association retrouvée dans 21 % des cas dans une série taïwanaise [9], dans 34 % des cas en Floride [10] et dans 25 % des cas à Mexico [11] dans une cohorte d’âge moyen 8,7 ans. Chez les enfants plus âgés, après 12 ans, le facteur de risque le plus fréquemment retrouvé est le port de lentilles [10]. Il s’agit de la deuxième étiologie après le traumatisme oculaire [9, 11].
Les maladies systémiques, la malnutrition principalement, font partie des principaux facteurs de risque de kératites microbiennes dans les pays en voie de développement [1, 11].
Il est nécessaire d’effectuer un examen complet avec un interrogatoire des parents à la recherche de l’histoire clinique et des antécédents de l’enfant. Un examen sous anesthésie générale sera effectué, en fonction de l’âge de l’enfant, afin de réaliser un examen clinique dans de bonnes conditions avec un prélèvement cornéen pour analyse microbiologique. Cet examen clinique permettra de réaliser un diagnostic positif de kératite infectieuse, un diagnostic de gravité, la recherche de facteur de risque et servira de référence pour le suivi de l’évolution clinique.
L’apparition des kératalgies est le plus souvent brutale. Un retard de prise en charge est possible du fait de l’absence de communication verbale chez les plus jeunes.
L’importance de la baisse d’acuité visuelle dépend de la localisation de la kératite infectieuse par rapport à l’axe optique, de l’inflammation intra-oculaire réactionnelle, de la présence de sécrétions et du larmoiement réflexe. Elle sera évaluée avec une échelle adaptée à l’âge après lavage abondant de l’oeil atteint et instillation d’oxybuprocaïne.
L’examen du segment antérieur permet de faire le diagnostic de kératite infectieuse et apprécie la gravité des lésions cornéennes et l’existence de complications. L’examen clinique initial peut objectiver un oedème palpébral, une hyperhémie conjonctivale, un cercle périkératique et un infiltrat localisé avec une ulcération épithéliale prenant la fluorescéine. Il précise : le nombre d’infiltrats, diffus ou localisés, leur localisation, leur dimension, leur forme et leur profondeur ; l’épaisseur cornéenne avec présence d’oedèmes ou non ; la visualisation de signes d’inflammation du segment antérieur ; la présence d’opacités anciennes et de néovaisseaux actifs ; l’écoulement de sécrétions, l’association avec une sclérite ou une endophtalmie.
Le diagnostic de gravité est nécessaire pour déterminer la prise en charge ambulatoire ou hospitalière. Une kératite infectieuse est dite sévère s’il y a la présence des critères cliniques suivants :
- abcès de plus de 2 mm de diamètre ;
- abcès de moins de 3 mm de l’axe visuel ;
- inflammation du segment antérieur (Tyndall supérieur à 1+) ;
- atteinte bilatérale ;
- absence d’amélioration ou aggravation à 24 heures de traitement ;
- risque de perforation cornéenne.
Des critères généraux de gravité doivent également être pris en compte : nourrisson, antécédents chirurgicaux, immunodépression, patient monophtalme et mauvaise observance du traitement.
Il est important de considérer chez les enfants, de moins de 2 ans, toute kératite bactérienne comme possiblement sévère. L’examen biomicroscopique permettra de nous orienter vers une kératite bactérienne (fig. 9-11), amibienne (fig. 9-12) ou mycotique en fonction des facteurs de risques présents et du type de lésion cornéenne.
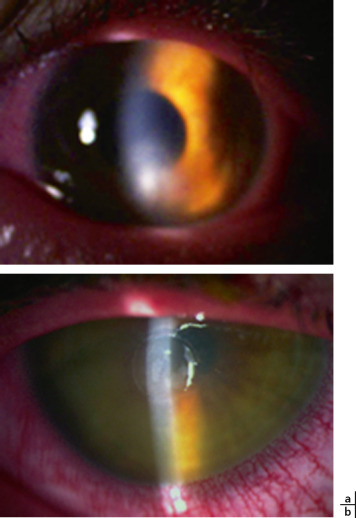
Fig. 9-11 Kératites bactériennes.
a. Aspect biomicroscopique d’un infiltrat cornéen de 1,5 mm de diamètre situé à 3 mm de l’axe visuel avec prise de fluorescéine positive chez un enfant de 8 ans sans facteur de risque retrouvé. b. Aspect biomicroscopique d’un infiltrat cornéen de 2,5 mm de diamètre central au niveau l’axe visuel avec prise de fluorescéine positive chez un enfant de 12 ans après traumatisme oculaire.
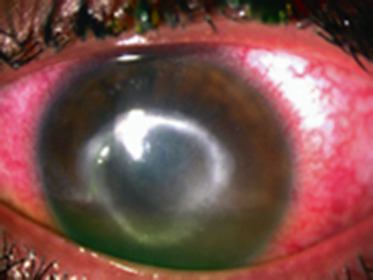
Fig. 9-12 Kératite amibienne.
Aspect biomicroscopique d’une kératite amibienne avec anneau immunitaire central chez une patiente de 14 ans porteuse de lentilles mensuelles.
Des photographies du segment antérieur peuvent éventuellement être réalisées et aideront au suivi objectif de l’évolution des lésions. Une surveillance clinique rapprochée sera impérative.
Le prélèvement cornéen est nécessaire pour un diagnostic microbiologique qui pourra permettre secondairement une adaptation thérapeutique. Chez les enfants, et notamment chez les nourrissons, le prélèvement sera effectué sous sédation ou anesthésie générale. Il sera effectué systématiquement en cas de kératite infectieuse sévère.
L’étude d’Al Otaibi et al., réalisée en Arabie saoudite en 2012 [8], retrouve sur 68 patients prélevés, de 4,5 ans d’âge moyen, 50 % des cultures positives. Ce taux de positivité des cultures après prélèvement cornéen varie selon les études de 48 à 87 % . Ces variations de résultats sur les prélèvements microbiologiques peuvent être expliquées par les différences des techniques d’analyse entre laboratoires et l’utilisation d’inhibiteurs topiques avant grattage cornéen.
La flore oculaire, palpébrale et conjonctivale, normale des nouveau-nés offre une protection contre les agents pathogènes externes. Elle comprend principalement des germes cutanés avec en majorité Staphylococcus epidermidis suivi par Lactobacillus, Bifidobacterium, Peptostreptococcus, Staphylococcus à coagulase négative et Propionibacterium [12]. Chez l’enfant de 2 à 6 ans, la flore conjonctivale est proche de la flore rhinopharyngée. Certains germes comme les streptocoques, en particulier Streptococcus pneumoniae ou Haemophilus influenzae, sont plus fréquents. Les corynébactéries sont plus abondantes chez l’adulte.
Les kératites infectieuses pédiatriques sont essentiellement bactériennes et les atteintes fongiques ou parasitaires sont rarement impliquées mais plus sévères.
Chez le nouveau-né, quatre facteurs favorisent la survenue d’une conjonctivite infectieuse pouvant se compliquer de kératite : une infection non traitée du tractus génital de la mère, les traumatismes obstétricaux et la rupture précoce des membranes ou un travail prolongé exposant le nouveau-né aux germes maternels. Les agents infectieux à l’origine de ces conjonctivites sont principalement Neisseria gonorrhoeae, Chlamydia trachomatis, le virus herpès simplex. Il existe également des kératites infectieuses néonatales reliées aux problèmes environnementaux hospitaliers dus au staphylocoque doré, Streptococcus pneumoniae, Haemophilus influenza, le streptocoque viridans ou les entérobactéries. Les kératites amibiennes sont associées au port de lentilles de contact et donc peu fréquentes chez le jeune enfant (fig. 9-12).
L’étude de Al Otaibi et al. [8] a rapporté, sur une série de 68 prélèvements, 67,8 % de cocci à Gram positif (Streptococcus pneumoniae, Streptococcus epidermidis et Streptococcus areus) et 32,2 % , de cocci à Gram négatif (Pseudomonas aeruginosa : 19,3 % , Haemophilus influenza et Moraxella catarrhalis). Ces résultats, incriminant majoritairement les germes cocci à Gram positif dans les kératites pédiatriques, sont concordant avec les autres études de la littérature [2, 11], contrairement à une étude effectuée en Floride [10], où Pseudomonas aeruginosa était le plus fréquemment observé (43,2 % ), mais l’analyse microbiologique était réalisée sur les lentilles de contact et non sur les prélèvements cornéens. Les agents pathogènes des kératites infectieuses varient donc en fonction de l’âge, de l’origine géographique des patients mais également de l’étiopathogénie :
- bacilles à Gram négatif chez les porteurs de lentilles de contact et cas de traumatismes ;
- Streptococcus pneumoniae et Haemophilus influenzae en présence d’une sténose lacrymale chronique ;
- Pseudomonas aeruginosa, Streptococcus aureus et bacilles à Gram négatif dans les cas de kératites bactériennes nosocomiales.
Les stratégies thérapeutiques actuelles ont pour objectif d’obtenir une destruction rapide de l’agent microbien impliqué tout en diminuant l’inflammation réactionnelle par instillation d’antiinflammatoires, essentiellement stéroïdiens, afin de limiter l’incidence des cicatrices stromales post-infectieuses potentiellement source de baisse d’acuité visuelle sévère. La plupart des ulcères infectieux survenant chez l’enfant sont traités avec succès par une thérapie topique seule [13]. Une kératite bactérienne constitue une urgence et une hospitalisation est nécessaire.
Le traitement de la kératite microbienne consiste en l’administration fréquente d’agents antimicrobiens agressifs, qui peut se compliquer d’une mauvaise observance et tolérance chez les enfants. Il n’y a pas actuellement de consensus concernant les indications des collyres antibiotiques dans le traitement des kératites bactériennes pédiatriques. Le traitement antibiotique par voie topique est débuté immédiatement après réalisation du prélèvement par grattage cornéen. Il est adapté à la gravité des lésions et à l’orientation suite de l’examen clinique. Les collyres renforcés en préparation hospitalière permettent d’obtenir de fortes concentrations cornéennes d’antibiotiques et sont indispensables dans le traitement des kératites bactériennes sévères. Les associations recommandées sont une bithérapie, associant généralement un aminoside (gentamycine 15 mg/ml) et une céphalosporine (céfazoline 50 mg/ml), ou une trithérapie associant carboxypénicilline (ticarcilline 6,6 mg/ml), aminoside (gentamycine 15 mg/ml) et glycopeptide (vancomycine 50 mg/ml). La supériorité d’une bi- ou trithérapie par antibiotiques fortifiés n’a pas été démontrée en comparaison à une monothérapie par fluoroquinolones de quatrième génération pour les kératites peu sévères [14]. Jeng et al. a rapporté 75 % de succès dans le traitement des ulcères de cornée par fluoroquinolone topique en monothérapie [13] en première intention, permettant ainsi de réduire la quantité et la fréquence d’instillation du traitement topique chez les enfants. L’utilisation de fluoroquinolones en monothérapie a reçu l’agrément de la Food and Drug Admnistration (FDA) mais des phénomènes de résistances bactériennes sont possibles. Dans tous les cas, une dose de charge par une instillation répétée toutes les 5 à 10 minutes la première heure de traitement permettra d’obtenir des concentrations cornéennes satisfaisantes précocement. La posologie sera ensuite d’une goutte par heure pendant 48 heures, puis une décroissance progressive sera effectuée en fonction de l’évolution clinique de l’atteinte cornéenne. Une injection sous-conjonctivale d’antibiotiques peut être proposée en cas de mauvaise observance des collyres, sans efficacité supérieure au traitement topique. L’utilisation de pommades antibiotiques est à éviter à la phase aiguë de l’infection pour ne pas diminuer la pénétration des collyres. Néanmoins, les pommades ont un temps de contact cornéen prolongé et sont particulièrement indiquées chez l’enfant ou en application au coucher.
Il est nécessaire de prescrire une association d’antiamibiens locaux : les diamidines aromatiques et les biguanides (fig. 9-12). Dans la classe des diamidines aromatiques on utilise le plus souvent l’hexamidine (Désomédine®), antiseptique actif sur les bactéries à Gram positif, et sur les amibes type Acanthamoeba. Dans la classe des biguanides, le plus utilisé est le polyhexaméthylène biguanide (PHBM) à 0,02 % en préparation magistrale, dont l’efficacité sur la forme kystique est supérieur à la Désomédine®.
La pénétration cornéenne des antifongiques topiques est faible. Néanmoins, la natamycine (5 % ), le fluconazole (0,5 % ), et l’amphotéricine B (0,25 % ) ont été rapportés comme efficaces dans le traitement des kératites fongiques chez l’enfant en association avec un traitement antifongique per os.
L’indication des corticostéroïdes reste controversée dans la prise en charge des kératites microbiennes. Ils diminuent la composante inflammatoire de la kératite bactérienne, mais peuvent retarder la guérison. Selon certains auteurs, les corticostéroïdes permettent de réduire les séquelles à type de cicatrice stromale en diminuant la taille de l’infiltrat [15] et la néovascularisation cornéenne. Blair et al. en 2011 [15] ont rapporté, dans une étude randomisée, une différence non statistiquement significative concernant la taille de l’infiltrat à 2,5 mois chez les patients traités par corticoïdes. Une amélioration de l’acuité de visuelle de deux lignes à 3 mois après instillation d’anti-inflammatoires stéroïdiens a également été rapportée chez ces patients [16]. Les corticoïdes topiques doivent être indiqués uniquement lorsque la prolifération microbienne est contrôlée et une surveillance clinique rapprochée possible. Ils peuvent être prescrits après un minimum de 48 heures de traitement antibiotique efficace.
Une prise en charge chirurgicale est rarement nécessaire en urgence. Elle peut consister en un débridement, seul ou en association avec une greffe de membrane amniotique. Il est possible aussi de proposer la réalisation d’un volet conjonctival de recouvrement. Il est parfois nécessaire de réaliser une kératoplastie lamellaire ou une kératoplastie transfixiante « à chaud » lors d’un risque de perforation ou d’une infection évolutive malgré un traitement anti-infectieux à forte dose. On favorisera la réalisation d’une greffe de membrane amniotique multicouche ou de la colle cyanoacrylate dans le cas d’une menace de perforation afin de différer l’éventualité d’une greffe de cornée [17]. À distance de l’épisode infectieux, une photokératectomie thérapeutique au laser Excimer (excited dimer), voire une kératoplastie lamellaire ou transfixiante pourront être proposées, en fonction de la profondeur et de la localisation de la taie cornéenne. Le taux d’intervention chirurgicale confondant les prises en charge en urgence et à distance a été rapporté entre 15,9 et 21 % [10, 18].
La surveillance des kératites infectieuses doit être quotidienne jusqu’à cicatrisation totale et éventuellement documentée par des photographies du segment antérieur pour une surveillance objective. La fréquence d’instillation des collyres et la durée du traitement sont adaptées en fonction de l’évolution clinique et de la nature du germe.
À distance de l’épisode infectieux, une surveillance de l’acuité visuelle avec correction optique totale est nécessaire. La correction optique sera réévaluée du fait d’un risque élevé d’astigmatisme secondaire. La prise en charge du risque d’amblyopie sera adaptée en fonction de l’âge.
Les facteurs et critères de gravité ainsi que les modalités de prise en charge des kératites infectieuses sont résumés dans le tableau 9-8.
[1] Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness : a global perspective. Bull World Health Organ 2001 ; 79 : 214-21.
[2] Ormerod LD, Hertzmark E, Gomez DS, et al. Epidemiology of microbial keratitis in southern California : a multivariate analysis. Ophthalmology 1986 ; 94 : 1322- 33.L.D.
[3] Ormerod AL, Murphree DS, Gomez DJ, et al. Microbial keratitis in children. Ophthalmology 1986 ; 93 : 449-55.
[4] Maurin JF, Renard JP, Ahmedou O, et al. Corneal blindness in tropical areas. Med Trop 1995 ; 55 : 445-9.
[5] Gonzales CA, Srinivasan M, Whitcher JP, et al. Incidence of corneal ulceration in Madurai District, South India. Ophthal Epidemol 1996 ; 3 : 159-66.
[6] Upadhyay MP, Karmacharya PC, Koirala S, et al. The Bhaktapur eye study : ocular trauma and antibiotic prophylaxis for the prevention of corneal ulceration in Nepal. Br J Ophthalmol 2001 ; 85 : 388-92.
[7] Erie JC, Nevitt MP, Hodge DO, et al. Incidence of corneal ulceration in a defined population from 1950–1988. Arch Ophthalmol 1991 ; 11 : 92-9.
[8] Al Otaibi AG, Allam K, Damri AJ, et al. Childhood microbial keratitis. J Ophthalmol 2012 ; 5 : 28-31.
[9] Hsiao CH, Yeung L, Ma DH, et al. Pediatric microbial keratitis in Taiwanese children : a review of hospital cases. Arch Ophthalmol 2007 ; 125 : 603-9.
[10] Clinch TE, Plamon FE, Robinson MJ, et al. Microbial keratitis in children. Am J Ophthalmol 1994 ; 117 : 65-71.
[11] Chirinos-Saldana P, Bautista de Lucio VM, Hernandez-Camarena JC, et al. Clinical and microbiological profile of infectious keratitis in children. BMC Ophthalmol 2013 ; 13 : 54.
[12] Asbell P, Stenon S. Ulcerative keratitis : survey of 30 years laboratory experience. Arch Ophthalmol 1982 ; 100 : 77-80.
[13] Jeng BH, McLeod SD. Microbial keratitis. Br J Ophthalmol 2003 ; 87 : 805-6.
[14] Shah VM, Tandon R, Dip NB, et al. Randomized clinical study for comparative evaluation of fourth-generation fluoroquinolones with the combination of fortified antibiotics in the treatment of bacterial corneal ulcers. Cornea 2010 ; 29 : 751-7.
[15] Blair J, Hodge W, Al-Ghamdi S, et al. Comparison of antibiotic-only and antibiotic- steroid combination treatment in corneal ulcer patients : double-blinded randomized clinical trial. Can J Ophthalmol 2011 ; 46 : 40-5.
[16] Srinivasan M, Lalitha P, Mahalakshmi R, et al. Corticosteroids for bacterial corneal ulcers. Br J Ophthalmol 2009 ; 93 : 198-202.
[17] Killingsworth DW, Stern GA, Driebe WT, et al. Results of therapeutic penetrating keratoplasty. Ophthalmology 1993 ; 100 : 534-41.
[18] Kunimoto DY, Sharma S, Reddy MK, et al. Microbial keratitis in children. Ophthalmology 1998 ; 105 : 252-7.
B. Mortemousque
Cette affection fait partie des réactions immuno-allergiques de la surface oculaire [1]. De mécanisme physiopathologique complexe, elle fait intervenir des mécanismes cellulaires et humoraux [1–4]. Il est important de comprendre que l’allergie (IgE médiée) n’est qu’un facteur aggravant ou déclenchant de la maladie, tout comme la sécheresse oculaire, la chaleur et les rayons UV, la corticothérapie au long cours : la kératoconjonctivite vernale est une pathologie complexe et multifactorielle.
Classiquement, elle débute dans l’enfance (80 % avant 10 ans). Avant l’âge de 20 ans, le sex-ratio est de 2 sur 4 en faveur des garçons. Après 20 ans, la maladie atteint autant les hommes que les femmes. Le plus souvent résolutive à l’adolescence, on observe dans 10 % des cas un passage à la chronicité donnant un tableau similaire à la kératoconjonctivite atopique.
Sa prévalence, sa gravité et son évolutivité sont variables selon les pays : la KCV est plus souvent sévère en Afrique et au Japon. L’évolution est le plus souvent perannuelle avec dans 77 % des cas des recrudescences en période de chaleur et d’ensoleillement à savoir de février à septembre.
La symptomatologie, bruyante lors des poussées, maximale le matin rendant le réveil de l’enfant difficile, est marquée par une sensation de corps étranger, plus ou moins associée à un prurit, un larmoiement et une photophobie intense. L’enfant peut présenter un blépharospasme (fig. 9-13a) avec très fréquemment une difficulté d’ouverture des yeux le matin (signant une atteinte cornéenne). Lors des épisodes aigus, des sécrétions muqueuses conjonctivales abondantes, épaisses peuvent prendre parfois un aspect de pseudo-membranes. L’enfant se présente sous une apparence caractéristique avec une tête dans la poitrine, des lunettes noires et une casquette (fig. 9-13b). Les symptômes lors des poussées inflammatoires sont invalidants, peuvent être responsables de véritables troubles du comportement et de retard scolaire. Ils ont un retentissement majeur sur la vie des enfants et sur celle des parents. La KCV peut entraîner une déscolarisation, des angoisses, des troubles du comportement qui justifient parfois un soutien psychologique.
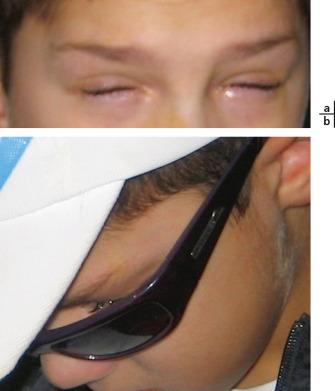
Fig. 9-13 Enfant présentant un blépharospasme avec larmoiements.
a. Difficulté d’ouverture des yeux encore plus important le matin. b. Apparence caractéristique – tête dans la poitrine, lunettes noires et casquette – d’un enfant en poussée de KCV.
d’un enfant en poussée de KCV. (Remerciements au Pr D. Denis.)
La KCV s’exprime selon plusieurs formes cliniques :
- la forme tarsale caractérisée par la présence de papilles géantes (diamètre > 1 mm) sur la conjonctive tarsale, le plus souvent supérieure (fig. 9-14a). Ces papilles peuvent être jointives réalisant un véritable pavage. Elles aboutissent à un épaississement palpébral responsable d’un ptosis (pseudo-ptosis). Dans les formes anciennes, la conjonctive apparaît comme fibrosée ;
- la forme limbique, qui peut être isolée ou associée à la forme palpébrale, est la plus fréquemment rencontrée chez les sujets mélanodermes dans sa forme limbique pure. On y observe un aspect de bourrelet gélatineux du limbe au sein duquel on peut individualiser des nodules blanc jaunâtre appelés grains de Trantas (fig. 9-14b) : amas d’éosinophiles responsables de la libération de protéases (eosinophil cationic protein [ECP] ++ ), de chémokines et d’autres médiateurs de l’inflammation.
Les atteintes cornéennes à type de kératites sont fréquentes dans les KCV et compliquent la maladie. Elles sont plus liées à la libération des médiateurs des cellules inflammatoires (éosinophiles ++) qu’au rôle mécanique des papilles géantes. Selon le degré de gravité de l’affection, ces lésions vont commencer par une kératite ponctuée superficielle (KPS) plus ou moins diffuse, responsable d’une altération de la fonction visuelle. Puis la KPS peut se compliquer d’un ulcère cornéen, dit vernal, souvent épithélial, ovalaire ou pentagonal, peu profond, avec des bords surélevés, peu douloureux et situés le plus souvent dans le tiers supérieur de la cornée (plaque vernale [fig. 9-14c] prenant la fluoroscéine [fig. 9-14d] et KPS [fig. 9-15]). Des cellules et du mucus à un stade de plus remplissent l’ulcération pour aboutir à une formation blanchâtre homogène, indurée, adhérente au fond de l’ulcère appelée plaque vernale. Cette plaque vernale va entretenir un processus inflammatoire local empêchant la cicatrisation épithéliale et laissant parfois des cicatrices cornéennes néovascularisées pouvant retentir sur la fonction visuelle. Elle peut évoluer vers la formation d’un pannus cornéen et est souvent le siège de surinfection, en particulier bactérienne.
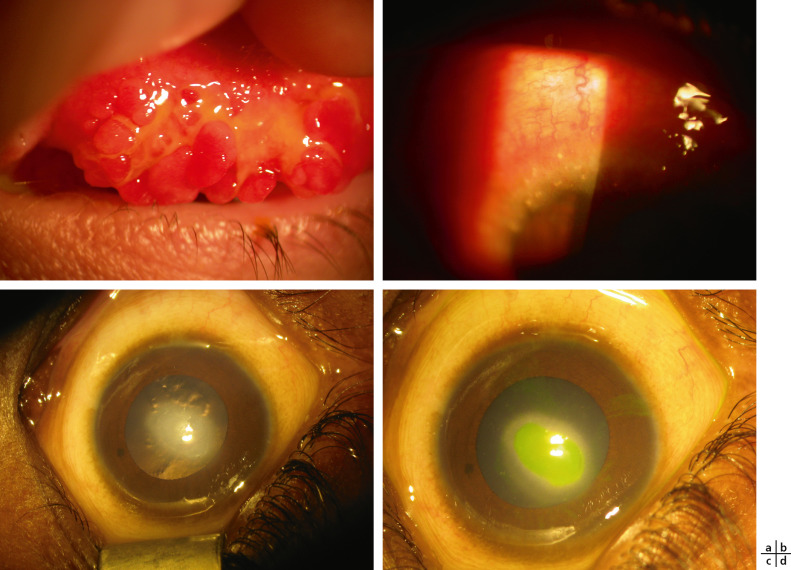
Fig. 9-14 Kératoconjonctivites vernales.
a. Forme tarsale caractérisée par la présence de papilles géantes (diamètre > 1 mm) sur la conjonctive tarsale, le plus souvent supérieure. b. Forme limbique avec aspect de bourrelet gélatineux du limbe avec nodules blanc jaunâtre appelés grains de Trantas. c, d. Plaque vernale, prenant la fluroscéine (d). (Fig. b : remerciements Dr S. Doan ; fig. c et d : remerciements Dr E. Bui Quoc.)
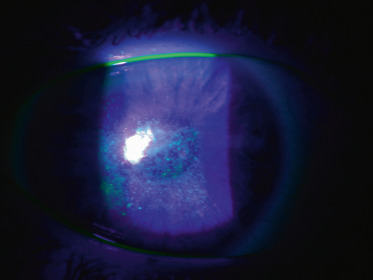
Fig. 9-15 Kératoconjonctivite vernale.
Kératite ponctuée superficielle plus ou moins diffuse responsable d’une altération de la fonction visuelle.
En dehors des poussées inflammatoires, ce traitement varie selon le degré d’inflammation et peut comprendre uniquement le port de verres filtrants les UV et d’une casquette associé à des lavages au sérum physiologique ou aux larmes artificielles non conservées. Dans les formes plus sévères, un stabilisateur de membrane en collyre sans conservateur suffit en général. Lors d’allergie associée, un collyre antihistaminique, parfois associé à un antihistaminique oral, peut être nécessaire. En cas de difficultés d’ouverture des yeux le matin, l’application au coucher de pommade (par exemple vitamine A pommade) doit être proposée.
Les verres solaires et la lubrification oculaire au sérum physiologique froid, sont indiqués. Les anti-inflammatoires non stéroïdiens (AINS) en collyre peuvent être prescrits mais sont souvent mal tolérés. En l’absence de kératite, les corticoïdes locaux ne sont pas indiqués. En cas de kératite ponctuée sévère confluente, des corticoïdes locaux doivent être prescrits mais en cure courte (2 semaines au maximum) avec des doses dégressives, et en surveillant le tonus oculaire, en particulier, chez l’enfant.
Un ulcère vernal [5] nécessite une corticothérapie locale utilisant un corticoïde puissant de type dexaméthasone ou bétaméthasone à forte dose (1 goutte 8 à 12 fois/j). Une couverture par un antibiotique local est souvent associée en raison du risque de surinfection. La surveillance de la fermeture de l’ulcère doit être quasi quotidienne, à la recherche d’une surinfection ou d’un amincissement cornéen. La prise en charge d’une plaque vernale nécessite un grattage chirurgical, sous anesthésie générale si l’enfant n’est pas coopérant, ou parfois à la lampe à fente sous anesthésie topique chez les plus grands. Ensuite, le traitement instauré est celui de l’ulcère vernal. Une membrane amniotique peut être utilisée dans les cas rebelles.
Dans les formes sévères corticodépendantes avec complication cornéenne, plusieurs traitements épargneurs de corticoïdes sont proposés, seuls ou en association. La ciclosporine en collyre, à concentration de 0,5 à 2 % , est certainement la meilleure indication aujourd’hui. Elle ne s’envisage que pendant les périodes inflammatoires et représente un agent épargneur de corticoïdes. Les immunosuppresseurs par voie systémique ne sont pas indiqués dans la KCV.
Afin de mieux identifier ces formes les plus graves de KCV qui sont les plus à risque de récurrences, d’ulcération de la cornée et de mauvais résultat visuel final, Sacchetti propose un système de notation en cinq sous-groupes, le sous-groupe 0 étant l’absence de symptômes et de traitement, le sous-groupe 5 étant une KPS diffuse ou un ulcère cornéen, avec haute dose de stéroïde topique. Un nombre plus élevé de rechutes et une gradation initiale plus élevée de KCV ont été les principaux facteurs de prédiction pour le plus mauvais résultat visuel [6].
Longtemps ignorée et encore sous-estimée aujourd’hui, la rosacée oculaire de l’enfant est une pathologie fréquente responsable de lésions cornéennes souvent non étiquetées [7]. Il s’agit d’une affection liée à un dysfonctionnement des glandes de Meibomius (DGM). L’expression clinique de la rosacée oculaire est extrêmement polymorphe, ce qui fait souvent errer le diagnostic. Le DGM est le plus souvent de type obstructif, plus rarement hyperproductif (séborrhée meibomienne), et parfois absent. Cependant, chez l’enfant les chalazions récidivants sont un bon signe d’appel et doivent être recherchés systématiquement. La blépharite est fréquente, postérieure ou mixte (associée à une blépharite séborrhéique), cependant les télangiectasies du bord libre et de la peau palpébrale ne sont pas toujours évidentes en particulier chez le jeune enfant. Outre la sécheresse évaporative chronique, des crises d’inflammation conjonctivale et/ou palpébrale sont classiques. En raison du polymorphisme clinique, son diagnostic chez l’enfant est probablement sous-évalué et la rosacée oculaire est considérée comme rare. Les signes cutanés sont très inconstants et volontiers fluctuants. Il s’agit le plus souvent de formes papulopustulaires, télangiectasiques et granulomateuses [8]. Chez l’adolescent, la rosacée et l’acné vulgaire peuvent coexister [9–11], mais dans cette dernière les comédons et les flushes sont absents.
Les signes oculaires d’appel sont les chalazions récidivants, une rougeur oculaire souvent unilatérale, une photophobie, plus rarement une baisse de vision [7]. L’association des deux premiers signes doit faire suspecter le diagnostic. Le temps de rupture du film lacrymal (break-up time [BUT]) est diminué. Le test de Schirmer est souvent normal mais peut être abaissé dans les formes évoluées. La vision est fluctuante, conséquence de l’instabilité lacrymale. Au niveau de la conjonctive, des papilles sont en général retrouvées orientant souvent à tort vers une allergie. Une hyperhémie conjonctivale bulbaire fluctuante est habituelle et prédominant dans la partie basse de l’oeil. Dans les formes évoluant depuis longtemps, une fibrose conjonctivale est classique, généralement discrète mais parfois à l’origine de symblépharons et d’un comblement des culs-de-sac conjonctivaux pouvant évoquer une pemphigoïde des muqueuses. Une conjonctivite phlycténulaire est plutôt l’apanage de la rosacée du sujet jeune. Épisclérite et sclérite nodulaire se rencontrent également dans la rosacée. Enfin, l’atteinte cornéenne inférieure est la plus fréquente et doit faire évoquer un DGM ou une blépharite. Une KPS est habituelle, banale et non spécifique. Une insuffisance limbique localisée se traduira par un pannus inférieur accompagné d’un fin pinceau néovasculaire qui est également extrêmement évocateur. Les complications immunologiques cornéennes sévères de la rosacée sont peu fréquentes (fig. 9-16), mais elles peuvent menacer la vision et l’intégrité cornéenne. Il existe deux types très caractéristiques, les ulcères ou infiltrats catarrhaux et les phlyctènes et phlycténules. Enfin, en l’absence de traitement adapté, des ulcérations voire des perforations cornéennes peuvent survenir.
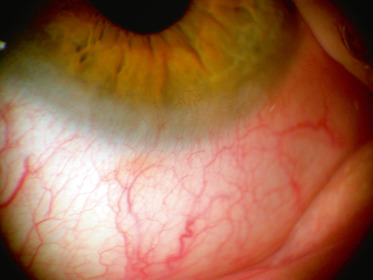
Comme dans tous les DGM, les soins des paupières (chauffages et massages) seront à proposer. Ils pourront s’accompagner d’instillation de collyre antibiotique (azythromycine). Dans les formes sévères et très inflammatoires, le collyre à la ciclosporine aura toute sa place. Pour ces deux traitements locaux, la règle sera une prescription de longue durée (3 mois minimum). Ainsi, en cas de phlyctènes, le traitement sera proposé jusqu’à leur disparition. Dans les formes associées à une atteinte cutanée, l’utilisation d’antibiotiques généraux sera préconisée (tétracyclines, macrolides et imidazolés) en tenant compte des restrictions d’utilisation liées à l’âge de l’enfant.
Initialement décrite par Thygeson en 1950, elle est présentée comme une KPS d’origine inconnue, bilatérale, mais pouvant être unilatérale (15 % des cas). Il s’agit d’un mécanisme immunologique sous-jacent probable, comme en témoignent son association génétique fréquente avec le human leukocyte antigen- DR3 (HLA-DR3) et sa sensibilité aux corticoïdes. Elle pourrait être déclenchée par un virus [12] ou par des atteintes autres de la surface oculaire (phototraumatisme, dénervation, micro-ulcérations mécaniques ou chimiques, port abusif de lentilles, réaction médicamenteuse). Par la suite, les phénomènes immunitaires seraient au premier plan.
L’atteinte cornéenne est isolée et peu bruyante (acuité visuelle quasi toujours conservée, photophobie, irritation). On retrouve des opacités intra-épithéliales ovales bien délimitées, blanc grisâtre, de topographie centrale, plus ou moins associées à des infiltrats sousépithéliaux transitoires qui ne laissent en règle générale pas de cicatrice (fig. 9-17). Classiquement, aucun signe conjonctival (ou hyperhémie minime), hypo-esthésie cornéenne ou adénopathie ne l’accompagne.
Chez l’enfant et le sujet jeune, la pathologie est le plus souvent bilatérale [13] et évolue de façon chronique sur un mode de poussées-rémissions, sur plusieurs années (entre 3 et 11 ans en moyenne, selon les études).
En dehors d’une baisse d’acuité majeure chez l’enfant ou de manifestations cliniques bruyantes, le traitement se compose essentiellement d’agents mouillants. Dans le cas d’atteinte sévère, et bien que connus pour prolonger la durée des poussées, les corticoïdes trouvent leur place dans le traitement de ces KPS pour prévenir les cicatrices cornéennes [14] tout comme le collyre à la ciclosporine en cas de cortico-dépendance.

Fig. 9-17 Kératite de Thygeson.
Opacités intra-épithéliales ovales bien délimitées, blanc grisâtre, de topographie centrale, plus ou moins associées à des infiltrats sous-épithéliaux.
[1] Mortemousque B. Conjonctivites allergiques. Encycl Méd Chir (Elsevier, Paris). Ophtalmologie, 21-130-E-10. 2013, 10(4) : 1-11.
[2] Leonardi A, Secchi AG. Vernal keratoconjunctivitis. Int Ophthalmol Clin 2003 ; 43 : 41-58.
[3] Motterle L, Diebld Y, Enriquez de la Salamanca A, et al. Altered expression of neurotransmitter receptors and neuromediators in vernal keratocunjunctivitis. Arch Ophthalmol 2006 ; 124(4) : 462-8.
[4] Bonini S, Bonini S, Lambiase A, et al. Vernal keratoconjunctivitis revisited : a case series of 195 patients with long-term followup. Ophthalmology 2000 ; 107 : 1157-63.
[5] Cameron JA. Shields ulcers and plaques of the cornea in vernal keratoconjunctivitis. Ophthalmology 1995 ; 102 : 985-93.
[6] Sacchetti M, Lambiase A, Mantelli F, et al. Tailored approach to the treatment of vernal keratoconjunctivitis. Ophthalmology 2010 ; 117 : 1294-9.
[7] Pisella PJ, Baudouin C, Hoang-Xuan T. Surface oculaire. Rapport de la Société française d’ophtalmologie 2015. Issy-les-Moulineaux : Elsevier Masson ; 2015, p. 213-29.
[8] Chamaillard M, Mortemousque B, Boralevi F, et al. Cutaneous and ocular signs of childhood rosacea. Arch Dermatol 2008 ; 144 : 167-71.
[9] Lacz NL, Schwartz RA. Rosacea in the pediatric population. Cutis 2004 ; 74 : 99-103.
[10] Erzurum SA, Feder RS, Greenwald MJ. Acne rosacea with keratitis in childhood. Arch Ophthalmol 1993 ; 111 : 228-30.
[11] Bourrat E, Rybojad M, Deplus S, et al. Rosacea with ocular involvement in a child. Ann Dermatol Venereol 1996 ; 123 : 664-5.
[12] Pepose JS, Holland GN, Wilhelmus KR. Ocular infection and immunity. St Louis : Mosby-Year Book ; 1996.
[13] Nagra PK, Rapuano CJ, Cohen EJ, Laibson PR. Thygeson’ssuperfcial punctate keratitis: ten years’experience. Ophthalmology 2004 ; 111 : 34-7.
[14] Fintelmann RE, Vastine DW, Bloomer MM, Margolis TP. Thygeson superfcial punctate keratitis and scarring. Cornea 2012 ; 31 : 1446-8.
M. Robert
Devant tout tableau de dystrophie de cornée (affection bilatérale, symétrique et lentement progressive) comme devant toute dystrophie rétinienne, la première étape est d’éliminer une cause systémique. L’infiltration de la cornée, lorsqu’elle résulte d’une cause systémique, est généralement le fait d’un processus métabolique. Le rôle de l’ophtalmopédiatre est ici capital, car des diagnostics de mucopolysaccharidoses (groupe de maladies dont le pronostic a été bouleversé ces dernières années par les traitements enzymatiques, à condition d’être administrés suffisamment tôt) sont régulièrement posés en consultation d’ophtalmologie pédiatrique devant une cornée opaque apparemment isolée, mais dont l’interrogatoire et la simple inspection du visage de l’enfant montrent qu’elle s’inscrit dans le cadre plus global d’une atteinte systémique. Ces maladies sont traitées dans le chapitre 26 consacré aux pathologies systémiques.
Les infections materno-foetales, à l’exception de la syphilis congénitale, sont une cause possible quoique rare de microcornée (toxoplasmose, rubéole et virus herpès simplex), mais ne sont pas responsables de « cicatrices » cornéennes.
La syphilis, même dans sa forme congénitale, demeure une « grande simulatrice » ; les manifestations ophtalmologiques sont extrêmement variées, souvent peu spécifiques, et peuvent toucher la totalité des structures de l’oeil et des voies visuelles et oculomotrices. L’atteinte de la cornée consiste en une kératite interstitielle, dont l’un des signes les plus spécifiques est la présence de néovaisseaux stromaux, souvent difficiles à visualiser chez un nouveau- né lorsqu’ils sont situés au sein d’une vaste opacité, même en utilisant une lampe à fente.
Les syndromes dysautonomiques sont à l’origine de kératopathies neurotrophiques sévères et exigent une surveillance et une prise en charge à partir de l’âge du diagnostic. Ils sont responsables avant tout d’une anesthésie congénitale de la cornée du groupe 2 de la classification de Rosenberg, avec d’autres affections ectodermiques ou mésenchymateuses (syndrome de Goldenhar, séquence de Möbius, associations VACTERL et MUCUS), à laquelle s’ajoutent les effets de l’hypo-innervation sympathique sur la prolifération épithéliale cornéenne ainsi que ceux de l’absence d’acétylcholine. Deux syndromes sont à connaître : le syndrome de Riley-Day (dysautonomie familiale) et le syndrome de Stüve-Wiedemann.
Le syndrome de Riley-Day affecte des enfants d’origine ashkénaze ; il s’agit d’une maladie récessive autosomique secondaire à des mutations dans le gène IKBKAP (9q31.3). La maladie se caractérise initialement par des difficultés d’alimentation du fait de troubles de la motricité digestive, associées à une alacrymie congénitale. La maladie atteint secondairement l’ensemble des systèmes pulmonaire, cardiaque, nerveux. L’absence de papilles fongiformes sur la langue est d’une aide précieuse à la suspicion diagnostique. L’atteinte cornéenne est sévère dans environ la moitié des cas.
Le syndrome de Stüve-Wiedemann est également transmis sur un mode récessif autosomique et résulte de mutations dans le gène LIFR (5p13.1). Il se caractérise par une dysplasie osseuse congénitale particulière associée à une dysautonomie. La plupart des enfants atteints meurent d’hyperthermie au cours de la première année de vie. Dans le cas contraire, la prise en charge des complications cornéennes, systématiques, est primordiale.
L’atteinte cornéenne des syndromes dysautonomiques consiste en un ulcère de cornée chronique. L’oeil reste ouvert en raison de l’absence de douleur. La situation est parfois aggravée par les traumatismes infligés par l’enfant à sa cornée insensible. Les substituts lacrymaux sont systématiques, la surveillance des parents et la réactivité des ophtalmologistes essentielle afin de prendre en charge les ulcères dès que possible ; les tarsorraphies latérales peuvent être utiles ; en cas d’opacité centrale majeure, les kératoplasties transfixiantes sont de très mauvais pronostic et doivent être contre-indiquées – sauf en cas de perforation cornéenne sur ulcère creusant –, tandis que les iridectomies chirurgicales à visée optique peuvent permettre le développement d’une fonction visuelle [1, 2].
[1] Injarie AM, Narang A, Idrees Z, et al. Ocular treatment of children with Stuve-Wiedemann syndrome. Cornea 2012 ; 31 : 269-72.
[2] Ramaesh K, Stokes J, Henry E, et al. Congenital corneal anesthesia. Surv Ophthalmol 2007 ; 52 : 50-60.
L. Vera, E. Bui Quoc
Le syndrome de Stevens-Johnson (SSJ) et le syndrome de Lyell (ou nécrolyse épidermique toxique) sont les variantes d’une même affection vésiculobulleuse [1, 2] d’origine le plus souvent médicamenteuse, parfois infectieuse. Ces deux variantes se différencient par le pourcentage de surface décollée/décollable : < 10 % pour le SSJ, > 30 % pour le syndrome de Lyell, entre 10 et 30 % pour les syndromes de recouvrement. Si la peau est le principal organe affecté, les muqueuses, notamment oculaires, sont fréquemment impliquées, aussi bien à la phase aiguë qu’à la phase chronique.
L’incidence, tous âges confondus, varie de 0,4 à 1,2 cas par million d’habitants et par an pour le syndrome de Lyell et de 1,2 à 6 cas par million d’habitants et par an pour le SSJ [3]. Le taux de mortalité est moindre pour les enfants que pour les adultes : il varie de 0 % [4] à 17 % [5, 6]. Les antibiotiques, anti-épileptiques et AINS sont les classes médicamenteuses les plus impliquées [7]. En revanche, la morbidité est plus importante chez les enfants : les muqueuses oculaires sont fréquemment impliquées, aussi bien à la phase aiguë, 93 % pour le SSJ [8] et 71 à 100 % pour le Lyell [8, 9], qu’à la phase chronique où l’incidence des séquelles est comprise entre 21 et 29 % [8, 10, 11].
La durée de la phase aiguë varie de 2 à 4 semaines [12]. Les tissus impliqués sont la conjonctive bulbaire et tarsale, la cornée, le bord libre et la peau des paupières. Le tableau clinique est variable, depuis la conjonctivite hyperhémique, présentation la plus fréquente, jusqu’au décollement de l’ensemble de l’épithélium de la surface oculaire. L’inflammation peut être majeure, et conduire à la formation de membranes, de symblépharons, de comblement des culs-de-sac, d’ulcération cornéenne et dans les cas les plus sévères de perforation. La meibomite est très fréquente.
Le premier examen doit avoir lieu dès que le diagnostic est établi. Le suivi doit comprendre une visite toutes les 24 à 48 heures, car le tableau clinique peut rapidement évoluer.
Le traitement au stade aigu doit être orienté vers la prophylaxie des infections, la prévention des synéchies et le contrôle de l’inflammation. Il repose essentiellement sur une lubrification efficace au moyen de gels dépourvus d’agents conservateurs et de pommade vitamine A. Les collyres antibiotiques ne sont utilisés qu’en cas de surinfection. Des lavages répétés au sérum physiologique aident à chasser les débris inflammatoires et les médiateurs de l’inflammation, présents sur la surface oculaire et les paupières, et diminuent le risque infectieux.
Les études menées sur des groupes d’enfants n’ont pas permis de démontrer l’efficacité des corticoïdes dans la prévention ou la sévérité de l’atteinte oculaire [13, 14], et les effets secondaires systémiques ne sont pas négligeables [13]. Les injections d’immunoglobulines par voie intraveineuse seraient encore moins efficaces chez les enfants que chez les adultes [15]. Les corticoïdes locaux sont également d’indication discutée [16].
Les manoeuvres instrumentales (conformateurs, libération mécanique des adhérences) sont largement pratiquées mais n’ont pas fait la preuve de leur efficacité dans la prévention de la formation des symblépharons.
La greffe de membrane amniotique, utilisée pour la première fois en 2002 chez deux enfants [17], doit être envisagée dès les premiers jours en cas de défect épithélial de la surface oculaire, d’ulcération du bord libre ou en présence de pseudo-membranes [18]. La membrane doit recouvrir la totalité de la surface oculaire jusqu’aux bords libres en suturant la membrane à la peau des paupières. La procédure doit éventuellement être répétée en cas d’ulcération ou d’inflammation sévère et persistante [19]. Chez l’enfant, cette technique est délicate, car elle nécessite une sédation profonde en unité de soins intensifs ou des grands brûlés, et l’anesthésie générale n’est pas envisageable en cas d’atteinte des muqueuses oropharyngées.
L’atteinte chronique est multifactorielle. L’inflammation chronique persistante et les ulcérations conjonctivales à répétition conduisent à la formation de cicatrices. Celles-ci sont responsables de diverses complications :
- syndrome sec par insuffisance lacrymale (oblitération des conduits lacrymaux excrétoires) et altération de la qualité des larmes (oblitération des glandes de Meibomius par kératinisation) ;
- comblement des culs-de-sac, formation de symblépharons empêchant parfois une fermeture correcte des paupières et limitant la motilité ;
- sténose des méats lacrymaux, entropion, trichiasis, distichiasis par fibrose du tarse.
Au niveau de la conjonctive, l’inflammation est responsable de métaplasie squameuse, allant de la simple diminution du nombre de cellules à mucus, à la kératinisation visible macroscopiquement. Elle peut conduire à une destruction des cellules souches, avec secondairement une néovascularisation et une conjonctivalisation de la cornée (fig. 9-18).
Ces différentes complications peuvent s’auto-entretenir, engendrant un cercle vicieux qu’il est difficile de rompre. Par exemple, les cicatrices palpébrales (kératinisation du bord libre, cils frotteurs) sont corrélées à la sévérité des complications cornéennes [20]. Ces microtraumatismes de la surface oculaire, répétés à chaque clignement, entretiennent à leur tour une inflammation.
Certains patients, en particulier les enfants, peuvent développer des inflammations conjonctivales récurrentes, en lien avec une vascularite à complexes immuns [21]. Dans une série de 55 enfants, 18 % ont présenté des récurrences jusqu’à 7 ans après le premier épisode [10].
L’inflammation au stade chronique peut être objectivée par différentes techniques d’imagerie, telles que la microscopie confocale in vivo : la présence de cellules dendritiformes est augmentée au niveau du stroma cornéen [22].

Fig. 9-18 Syndrome de Lyell.
a, b. Néovascularisation cornéenne. c. Stade séquellaire après perforation au stade aigu.
Le traitement médical vise à contrôler l’inflammation, tout en limitant la toxicité des traitements. Le traitement de la sécheresse oculaire est non spécifique : substituts lacrymaux sans conservateur, mesures environnementales (humidificateurs, lunettes à chambre humide, etc.), arrêt si possible des traitements généraux favorisant la sécheresse. Le collyre à la ciclosporine pourrait être bénéfique aux patients qui ne présentent pas d’intolérance [23] et le sérum autologue peut être utile dans les défects épithéliaux persistants.
Les verres scléraux ont plusieurs avantages [24] : réfractif (correction des astigmatismes irréguliers) ; protection mécanique contre l’irritation des cils et des paupières kératinisées ; maintien d’un réservoir liquidien permanent devant la cornée. Ces lentilles améliorent de façon considérable la photophobie chez certains patients et donc leur qualité de vie [22].
La levée des symblépharons est indispensable avant toute reconstruction de la surface oculaire. Elle repose sur l’exérèse de la conjonctive pathologique et son remplacement soit par une greffe de muqueuse buccale [25], soit par une greffe de membrane amniotique. Lorsque la surface oculaire est détruite, il n’est pas possible de réaliser une greffe de cornée car l’épithélium pathologique du receveur recouvre le greffon et l’opacifie rapidement en l’absence de cellules souches de cornée. Il faut reconstituer chirurgicalement l’épithélium cornéen en transplantant des cellules souches. L’atteinte étant le plus souvent bilatérale, l’autogreffe limbique est souvent exclue [26]. Les résultats de l’allogreffe kératolimbique sont peu encourageants : rejets fréquents, glaucome, perte du greffon, complications iatrogènes liées à la nécessité d’une immunosuppression par voie générale [27].
En cas de cécité bilatérale liée à une opacification cornéenne totale, la pose chirurgicale d’une kérathoprothèse doit être envisagée avec une grande prudence chez un enfant, car son pronostic à long terme est très réservé [28].
Les syndromes de Stevens-Johnson et de Lyell sont des affections cutanéomuqueuses le plus souvent d’origine médicamenteuse. Elles sont rares et graves. Elles se caractérisent par une nécrose étendue de l’épiderme et des muqueuses. Les séquelles ophtalmologiques peuvent être dramatiques et sont de survenue imprévisible, imposant un suivi rapproché.
Le taux de mortalité est moins élevé que chez l’adulte mais plus de la moitié des enfants sont atteints de complications à long terme.
[1] Stevens AM, Johnson FC. A new eruptive fever associated with stomatitis and ophtalmia. Report of two cases in children. Am J Dis Child 1922 ; 24 : 526-33.
[2] Lyell A. Toxic epidermal necrolysis : an eruption resembling scalding of the skin. Br J Dermatol 1956 ; 68 : 355-61.
[3] Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med 1994 ; 331 : 1272-85.
[4] Hamilton GM, Fish J. Pediatric toxic epidermal necrolysis : an institutional review of patients admitted to an intensive care unit. J Burn Care Res 2013 ; 34 : e351-e358.
[5] Forman R, Koren G, Shear NH. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis in children: a review of 10 years’experience. Drug Saf 2002 ; 25 : 965-72.
[6] Singalavanija S, Limpongsanurak W. Stevens-Johnson syndrome in Thai children : a 29-year study. J Med Assoc Thai 2011 ; 94(Suppl 3) : S85-S90.
[7] Letko E, Papaliodis DN, Papaliodis GN, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis : a review of the literature. Ann Allergy Asthma Immunol 2005 ; 94 : 419-36. quiz 436-8, 456.
[8] Prendiville JS, Hebert AA, Greenwald MJ, Esterly NB. Management of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. J Pediatr 1989 ; 115 : 881-7.
[9] Jones WG, Halebian P, Madden M, et al. Drug-induced toxic epidermal necrolysis in children. J Pediatr Surg 1989 ; 24 : 167-70.
[10] Finkelstein Y, Soon GS, Acuna P, et al. Recurrence and outcomes of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Pediatrics 2011 ; 128 : 723-8.
[11] Ferrandiz-Pulido C, Garcia-Fernandez D, Dominguez-Sampedro P, Garcia-Patos V. Stevens- Johnson syndrome and toxic epidermal necrolysis in children : a review of the experience with paediatric patients in a university hospital. J Eur Acad Dermatol Venereol 2011 ; 25 1153-9.
[12] Revuz J, Pendo D, Roujeau JC, et al. Toxic epidermal necrolysis : clinical findings and prognosis factors in 87 patients. Arch Dermatol 1987 ; 123 : 1160-5.
[13] Rasmussen JE. Erythema multiforme in children. Response to treatment with systemic corticosteroids. Br J Dermatol 1976 ; 95 : 181-6.
[14] Leaute-Labreze C, Lamireau T, Chawki D, et al. Diagnosis, classification, and management of erythema multiforme and Stevens-Johnson syndrome. Arch Dis Child 2000 ; 83 : 347-52.
[15] Kim KH, Park SW, Kim MK, Wee WR. Effect of age and early intervention with a systemic steroid, intravenous immunoglobulin or amniotic membrane transplantation on the ocular outcomes of patients with Stevens-Johnson syndrome. Korean J Ophthalmol 2013 ; 27 : 331-40.
[16] Sotozono C, Ueta M, Koizumi N, et al. Diagnosis and treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis with ocular complications. Ophthalmology 2009 ; 116 : 685-90.
[17] John T, Foulks GN, John ME, et al. Amniotic membrane in the surgical management of acute toxic epidermal necrolysis. Ophthalmology 2002 ; 109 : 351-60
[18] Gregory DG. Treatment of acute Stevens-Johnson syndrome and toxic epidermal necrolysis using amniotic membrane : a review of 10 consecutive cases. Ophthalmology 2011 ; 118 :908-14.
[19] Hsu M, Jayaram A, Verner R, et al. Indications and outcomes of amniotic membrane transplantation in the management of acute Stevens-Johnson syndrome and toxic epidermal necrolysis : a case-control study. Cornea 2012 ; 31 : 1394-402.
[20] Di Pascuale MA, Espana EM, Liu DT, et al. Correlation of corneal complications with eyelid cicatricial pathologies in patients with Stevens-Johnson and toxic epidermal necrolysis syndrome. Ophthalmology 2005 ; 112 : 904-12.
[21] Foster CS, Fong LP, Azar D, et al. Episodic conjunctival inflammation after Stevens- Johnson Syndrome. Ophthalmology 1988 ; 95 : 453-62.
[22] Vera LS, Gueudry J, Delcampe A, et al. In vivo confocal microscopic evaluation of corneal changes in chronic Stevens-Johnson syndrome and toxic epidermal necrolysis. Cornea 2009 ; 28 : 401-7.
[23] Prabhasawat P, Tesavibul N, Karnchanachetanee C, Kasemson S. Efficacy of cyclosporine 0.05 % eye drops in Stevens Johnson syndrome with chronic dry eye. J Ocul Pharmacol Ther 2013 ; 29 : 372-7.
[24] Tougeron-Brousseau B, Delcampe A, Gueudry J, et al. Vision-related function after scleral lens fitting in ocular complications of Stevens-Johnson syndrome and toxic epidermal necrolysis. Am J Ophthalmol 2009 ; 148 : 852-859.e2
[25] Iyer G, Pillai VS, Srinivasan B, et al. Mucous membrane grafting for lid margin keratinization in Stevens-Johnson syndrome : results. Cornea 2010 ; 29 : 146-51.
[26] Kenyon KR, Tseng SC. Limbal autograft transplantation for ocular surface disorders. Ophthalmology 1989 ; 96 :709-22. Discussion 722-3.
[27] Iyer G, Srinivasan B, Agarwal S, et al. Treatment modalities and clinical outcomes in ocular sequelae of Stevens-Johnson syndrome over 25 years- a paradigm shift. Cornea 2016 ; 35 : 46-50.
[28] Alexander JK, Basak SK, Padilla MD, et al. International outcomes of the Boston type 1 keratoprosthesis in Stevens-Johnson syndrome. Cornea 2015 ; 34 : 1387-94.
N. Stolowy, L. Hoffart
Les kératopathies iatrogènes représentent une part non négligeable des atteintes cornéennes chez l’enfant. C’est pourquoi la surveillance des traitements oculaires topiques et systémiques doit être rigoureuse afin d’éviter l’apparition d’une iatrogénicité.
Vingt-quatre pour cent des pathologies de la surface oculaire seraient consécutives à une iatrogénie. Les mécanismes impliqués sont multiples et peuvent être associés entre eux [1, 2].
Les mécanismes immunologiques représentent 3 à 10 % des réactions d’intolérance [1]. Ces mécanismes sont surtout liés à une réaction d’hypersensibilité retardée de type IV et plus rarement à une hypersensibilité immédiate de type I [1, 2]. Cliniquement, les atteintes d’origine immunologique se manifestent essentiellement par un eczéma de contact palpébral. Ces réactions sont majorées chez le patient atopique [2].
La toxicité d’un collyre peut être liée à un effet cytotoxique direct, à la nature de son pH, à son osmolarité ou à une réaction de photosensibilisation [1]. La toxicité directe du collyre peut entraîner une nécrose cellulaire directe ou conduire, par des activations de messagers intracellulaires en cascade, à une apoptose et une réaction inflammatoire chronique d’origine neurogène. Cette toxicité est souvent cumulative, c’est-à-dire qu’elle apparaît au-delà d’une certaine dose qui est atteinte souvent après plusieurs mois voire plusieurs années ; la ciclosporine A collyre aurait une toxicité cornéenne endothéliale dose-dépendante qui a été retrouvée chez l’animal [3], mais qui n’a jamais été démontrée chez l’homme.
Certains médicaments topiques, notamment les collyres anti-infectieux (aminosides, fluoroquinolones, antiviraux telle l’idoxuridine) et corticoïdes, entraînent des modifications de la flore microbienne saprophyte de la surface oculaire [1] et entraînent par ce biais une toxicité locale par modification du film lacrymal et une sélection de certains germes accroissant ainsi l’incidence d’infections bactériennes ou fongiques.
- Kératite ponctuée superficielle : elle est typiquement inférieure ou nasale initialement et peut être diffuse en cas d’atteinte prolongée ou d’emblée d’atteinte majeure (fig. 9-19).
- Kératite filamenteuse.
- Opacités nummulaires.
- Ulcère marginal immuno-allergique.
- Ulcère toxique étendu qui peut prendre une forme pseudodendritique.
- Kératite neurotrophique : l’hypoesthésie cornéenne ou l’anesthésie cornéenne, induites par les collyres anesthésiants, entraînent une diminution des sécrétions lacrymales et du clignement, et globalement une diminution de la qualité du film lacrymal. Cela aboutit à une diminution de la régénération cellulaire cornéenne. De plus, les mécanismes mis en jeu dans ce défaut de cicatrisation cornéenne aboutissent à une stimulation des métalloprotéases qui accélèrent la dégradation des protéines de la matrice extracellulaire [4]. Cliniquement, il faut impérativement rechercher une hypoesthésie cornéenne. À l’examen biomicroscopique, une kératite ponctuée dans l’espace interpalpébral ainsi qu’un oedème cornéen modéré peuvent s’observer. L’atteinte cornéenne peut se présenter sous la forme d’une large ulcération épithéliale aux bords enroulés, de forme ovale, prédominant en inférieur ou dans l’espace interpalpébral, avec un oedème stromal modéré sousjacent. Dans les cas plus sévères, un ulcère cornéen stromal survient et peut aller jusqu’à la perforation, en particulier dans les cas de surinfection [5].
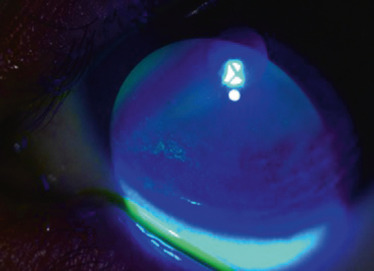
Fig. 9-19 Kératite ponctuée superficielle inféronasale après introduction d’azithromycine topique pour le traitement d’une blépharite chez un enfant de 5 ans.
- Atteinte conjonctivale : un chémosis et un ulcère conjonctival peuvent être présents. On peut aussi observer des papilles conjonctivales ou des érosions conjonctivales rondes qui prédominent au niveau de la conjonctive nasale et le long de la rivière lacrymale [6]. Certains collyres peuvent entraîner l’apparition d’une conjonctivite fibrosante avec une fibrose sous-conjonctivale pouvant aller jusqu’au symblépharon voire à l’ankyloblépharon : il s’agit en particulier des collyres antiglaucomateux dans 25 à 30 % des cas [6] et des antiviraux locaux [6].
- Atteinte palpébrale : il peut s’agir de lésions d’urticaire, témoignant d’une réaction d’hypersensibilité de type I, sous forme de plaques érythémateuses bien limitées, prurigineuses et fugaces. Il s’agit le plus fréquemment de lésions d’eczéma érythématocroûteuses qui témoignent d’une manifestation d’hypersensibilité de type IV avec des lésions érythémateuses mal limitées présentant un aspect sec et desquamatif, pouvant évoluer vers une lichénification donnant un aspect sec et quadrillé aux paupières [7]. Tous les collyres et pommades ophtalmologiques, y compris les collyres corticoïdes ou antihistaminiques, peuvent entraîner un eczéma de contact ou réaction d’hypersensibilité de type IV mais les collyres le plus souvent en cause sont les collyres bêtabloquants [7].
Les collyres hypotonisants sont des traitements au long cours, dont la toxicité est majorée chez les enfants car ils ont été débutés précocement. Ils entraînent une toxicité aiguë par un mécanisme allergique ou une toxicité directe ou cumulative pouvant survenir après plusieurs mois d’instillation [1]. La toxicité est plus importante en cas d’association de plusieurs collyres hypotonisants [1]. La toxicité des collyres antiglaucomateux est en partie causée par la toxicité directe ou indirecte des conservateurs, en particulier du chlorure de benzalkonium (voir plus loin).
Tous les collyres antibiotiques peuvent entraîner une toxicité cornéenne ; les lésions sont peu spécifiques : kératite ponctuée superficielle, kératite filamenteuse. Certains collyres antibiotiques présentent une atteinte plus caractéristique : les aminosides (néomycine, gentamicine, tobramycine) peuvent entraîner une nécrose conjonctivale [8], les fluoroquinolones (norfloxacine, ciprofloxacine, ofloxacine) peuvent générer des dépôts cornéens après instillations répétées [5].
Les lésions secondaires aux collyres antiviraux sont peu spécifiques : kératite ponctuée superficielle, retard de cicatrisation. Il est difficile de différencier les lésions initiales des lésions iatrogènes ; une toxicité locale doit être évoquée en cas de nonamélioration après une durée de traitement adaptée. Certains collyres antiviraux ont une toxicité plus importante, notamment les antiviraux de première génération tels que l’idoxuridine et la vidarabine ; ils entraînent des kératites stromales, des retards de cicatrisation importants et parfois des occlusions des points lacrymaux [6].
Les collyres corticoïdes sont responsables d’un retard de cicatrisation épithéliale cornéenne (parfois jusqu’à 30 % d’allongement du temps de cicatrisation) [6]. Ils peuvent aggraver l’évolution d’une kératite herpétique ou fongique.
Ces collyres entraînent l’apparition de kératites ponctuées superficielles, d’ulcérations cornéennes et parfois de perforations cornéennes. Leur toxicité est d’autant plus importante que l’utilisation est prolongée. Le diclofénac collyre présente une toxicité accrue par rapport aux autres collyres AINS [9]. Cette toxicité est corrélée au temps d’exposition et à la dose.
Les collyres anesthésiques (oxybuprocaïne, tétracaïne) sont responsables de kératites neurotrophiques iatrogènes (voir plus haut). L’anesthésie cornéenne diminue la fréquence du clignement, inhibe la sécrétion lacrymale et empêche ainsi la stimulation de la cicatrisation épithéliale.
La phényléphrine peut présenter une toxicité pour l’endothélium cornéen en cas d’injection intra-oculaire [10].
Les conservateurs comportent les ammoniums quaternaires dont le principal représentant est le chlorure de benzalkonium suivi par les dérivés organomercuriels (phénylmercure, mercurobutol et mercurothiolate sodique), les amidines (dont la chlorhexidine), les alcools (le chlorobutanol et le phényléthanol), les parabens, les complexes oxychlorés, le perborate de sodium et le système Sof- Zia® composé d’acide borique, de propylène glycol, de sorbitol et de chlorure de zinc.
Ils sont responsables de l’apparition des signes d’hypersensibilité tels que les sensations de prurit, de brûlures, de grains de sable, d’une hyperhémie conjonctivale. Ils peuvent entraîner une sécheresse oculaire, des ulcérations cornéennes [11, 12].
Les traitements systémiques entraînent une toxicité cornéenne par le biais d’une diffusion par le film lacrymal ou l’humeur aqueuse voire la circulation périlimbique [13].
Il s’agit de dépôts épithéliaux en forme de fougère qui apparaissent dans la partie inférieure de la cornée en laissant un espace de cornée claire périphérique. Ces dépôts ne sont pas responsables d’une baisse d’acuité visuelle. La cornea verticillata est le plus fréquemment liée à un traitement prolongé par antipaludéens de synthèse ou par amiodarone. L’hydroxychloroquine est contre-indiquée chez les enfants de moins de 6 ans mais peut être prescrite dans les cas de lupus pédiatrique. Un traitement par amiodarone peut être prescrit par les cardiopédiatres dans les cas de trouble du rythme chez l’enfant.
Les isotrétinoïdes (Roaccutane®) sont administrés dans les cas d’acné sévère ou de certaines dermatoses génétiques. Ils entraînent des blépharoconjonctivites, une sécheresse oculaire, et peuvent également entraîner des dépôts grisâtres au niveau du stroma cornéen [13].
La prise de chlorpromazine (Largactil®) au long cours peut entraîner l’apparition de dépôts cornéens brun jaunâtre, de taille réduite, diffus au niveau de l’endothélium cornéen et du stroma profond.
L’argyrose est causée par l’exposition à l’argent et provoque une coloration grisâtre de la cornée (dépôts gris au niveau de la membrane de Descemet) ou de la conjonctive. Cette atteinte peut être due à une exposition environnementale ou peut être iatrogène ; l’argent était utilisé pour ses propriétés antiseptiques et est encore utilisé dans certaines médecines traditionnelles [14].
On peut observer chez les patients traités au long cours par sels d’or des dépôts stromaux gris ou violets. Les sels d’or ou allochrysine sont indiqués chez les enfants dans le traitement de fond de l’arthrite chronique juvénile.
L’exposition orbitaire et des annexes à une radiothérapie externe ou curiethérapie peut entraîner un syndrome sec sévère. Elle peut également être responsable d’une kératite neurotrophique. La radiothérapie est encore actuellement indiquée dans le traitement des rétinoblastomes avec de nombreux effets indésirables [15].
- Kératite ponctuée superficielle : il s’agit de l’atteinte cornéenne liée aux lentilles de contact la plus fréquente. Sa localisation à 3 heures et 9 heures oriente vers un syndrome sec associé ou un clignement palpébral incomplet [5].
- Syndrome de la lentille serrée : on peut observer une indentation et un marquage par la fluorescéine de l’épithélium conjonctival en anneau autour de la cornée [5].
- Hypoxie aiguë : on observe des microkystes épithéliaux et des macro-érosions douloureuses [5].
- Hypoxie chronique : on constate une néovascularisation cornéenne et une kératopathie lipidique peut être associée [5].
Son mécanisme est proche de celle de la kératite marginale. Elle se présente sous la forme d’infiltrats marginaux sans lésion épithéliale ou avec des lésions minimes. Elle est accompagnée d’une hyperhémie conjonctivale minime.
Elle est causée par une atteinte chimique aiguë secondaire à un port de lentilles sans neutralisation du peroxyde d’hydrogène, contenu dans les solutions de nettoyage, ou par une toxicité chronique des agents désinfectants comme le thiomersal ou le chlorure de benzalkonium [5]. Elle se manifeste par une douleur aiguë, une rougeur et un chémosis dès la mise en place de la lentille. Elle peut aboutir dans les cas chroniques à une néovascularisation cornéenne et à des cicatrices cornéennes et limbiques.
Les kératopathies iatrogènes chez l’enfant peuvent sérieusement engager le pronostic visuel, en particulier si leur cause n’est pas identifiée rapidement et que la prise en charge est retardée.
Les effets indésirables des traitements oculaires topiques ne doivent pas être sous-estimés. Leur prise en charge repose avant tout sur leur diagnostic précoce et l’arrêt ou la diminution du traitement en cause. L’utilisation de collyres sans conservateur est à privilégier.
Les effets indésirables des traitements systémiques doivent être recherchés par une surveillance rigoureuse. Les kératites liées au port de lentilles de contact peuvent également entraîner des complications qui engagent le pronostic fonctionnel et ne doivent pas être méconnues.
[1] Bresson-Dumont H. Tolérance locale des médications antiglaucomateuses un problème sous-estimé. Bull Soc Belge Ophtalmol 2010 ; (215) : 47-53.
[2] Bartlett JD, Jaanus SD. Clinical ocular pharmacology. Boston : Butterworth-Heinemann ; 1995.
[3] Robert PY, Leconte V, Olivé C, et al. Collyre à la ciclosporine A : fabrication, toxicité, pharmacocinétique et indications en l’an 2000. J Fr Ophtalmol 2001 ; 24 : 527-35.
[4] Gabison EE, Huet E, Baudouin C, Menashi S. Direct epithelial-stromal interaction in corneal wound healing : role of EMMPRIN/CD147 in MMPs induction and beyond. Prog Retin Eye Res 2009 ; 28 : 19-33.
[5] Bowling B. Ed. Kanski’s Clinical ophthalmology, 8th ed. Elsevier ; 2016.
[6] Baudoin C. Pathologie iatrogène de la conjonctive et de la cornée. Rev Fr Allergol Immunol Clin 2002 ; 42 : 79-87.
[7] Collet E, Castelain M. Dermatites de contact des paupières. Ann Dermatol Venereol 2002 ;129 : 928-30.
[8] Davison CR, Tuft SJ, Dart JK. Conjunctival necrosis after administration of topical fortified aminoglycosides. Am J Ophtalmol 1991 ; 111 : 690-3.
[9] Lee JS, Kim YH, Park YM. The toxicity of nonsteroidal anti-inflammatory eye drops against human corneal epithelial cells in vitro. J Korean Med Sci 2015 ; 30 : 1856-64.
[10] Hong JW, Park JH, Kim ES, et al. Effect of intracameral injection of bisulfite-containing phenylephrine on rabbit corneal endothelium. Cornea 2015 ; 34 : 460-3.
[11] Vaede D, Baudouin C, Warnet JM, Brignole-Baudouin F. Les conservateurs des collyres : vers une prise de conscience de leur toxicité. J Fr Ophtalmol 2010 ; 33 : 505- 24.
[12] Baudouin C, Labbé A, Liang H, et al. Preservatives in eyedrops : the good, the bad and the ugly. Prog Retin Eye Res 2010 ; 29 : 312-34.
[13] Ravet O. La toxicité médicamenteuse sur la cornée. Bull Soc Belge Ophtalmol 2007 ; (304) : 154-4.
[14] Niezborala M. Toxicité des métaux précieux : or, argent, platine, palladium. Encycl Méd Chir (Elsevier, Paris). Pathologie professionnelle et de l’environnement, 16-003-M- 60. 1996
[15] Frikha H, Chaari N, Ben Nasr C, Ayed S. Place de la radiothérapie dans le traitement du rétinoblastome : à propos de 40 cas. Cancer/Radiothérapie 2009 ; 13 : 30-6.
H. Guigue, L. Hoffart
En zone tropicale, les déficits en micronutriments essentiels sont étroitement intriqués avec les carences nutritionnelles en énergie et en protides. Il en est ainsi des avitaminoses, en particulier de l’avitaminose A due à une carence en vitamine A ou rétinol. L’avitaminose A concerne 6 à 7 millions d’enfants par an [1], dont 500 000 deviennent aveugles en l’absence de traitement. Il s’agit de la première cause de cécité chez l’enfant dans le monde [2].
La carence en vitamine A est plus fréquente entre 6 et 36 mois au moment du sevrage. Les régions les plus concernées sont l’Asie, depuis l’Afghanistan jusqu’aux Philippines, l’Afrique sahélienne et l’Afrique orientale, l’Amérique centrale et le Brésil.
La vitamine A est retrouvée sous forme d’ester de rétinol dans les produits d’origine animale (foie, oeuf) et les produits laitiers. La provitamine A, ou bêtacarotène, est retrouvée dans les végétaux : fruits et légumes à chair orangée (carotte, melon, orange, abricot, mangue, papaye) et légumes à feuilles vertes. Les besoins journaliers en vitamine A chez l’enfant sont de 1 500 UI dont au moins 60 % doivent être apportés sous forme de bêtacarotène [3].
La vitamine A ingérée est libérée dans l’estomac puis l’intestin grêle. Le rétinol atteint ensuite le foie par le système porte, le foie contient en effet 90 % de la vitamine A de l’organisme. À partir du foie, le rétinol est ensuite libéré dans le sang où il se lie à la retinol binding protein (RBP). L’élimination de la vitamine A se fait dans les urines et les selles. Elle a une demi-vie de 4 à 5 mois.
La vitamine A joue un rôle important dans de nombreuses fonctions physiologiques :
- fonction visuelle : le rétinol est un des constituants de la rhodopsine, protéine photosensible indispensable à la fonction des bâtonnets et donc à l’adaptation à l’obscurité ;
- trophicité des épithéliums conjonctivaux et cutanés : la carence en vitamine A entraîne une incapacité des cellules caliciformes conjonctivales à produire du mucus ce qui entraîne une xérose conjonctivale et cornéenne ;
- trophicité des épithéliums du tube digestif et des voies respiratoires : l’avitaminose A est une cause importante de morbimortalité dans les pays en voie de développement en raison de la fréquence des gastroentérites et des bronchopneumonies qu’elle entraîne ;
- les défenses immunitaires : sensibilité accrue aux infections virales (rougeole, diarrhées) et aux complications pulmonaires.
Les étiologies de la carence en vitamine A peuvent être liées à :
- une malnutrition avec un apport alimentaire insuffisant en vitamine A. La carence alimentaire en vitamine A est exceptionnelle dans les pays industrialisés, elle est observée essentiellement dans les pays en voie de développement ;
- une malabsorption secondaire à une cholestase, une atrésie des voies biliaires, une maladie coeliaque, une résection intestinale, des parasitoses digestives, une pathologie inflammatoire du tube digestif ou une diarrhée chronique ;
- rougeole : besoin accru en vitamine A pendant et après la maladie.
L’avitaminose A est longtemps asymptomatique et peut se manifester par des signes cliniques ophtalmologiques, cutanés ou généraux.
L’atteinte ophtalmologique de l’avitaminose A, généralement bilatérale et symétrique, entraîne initialement une héméralopie puis des lésions conjonctivales ou cornéennes, accompagnées de taches de Bitot, pathognomoniques et réversibles, réalisant la xérophtalmie qui précède la survenue des lésions cornéennes irréversibles conduisant à la cécité. La conjonction d’une rougeole ou d’une malnutrition avec une avitaminose A peut entraîner une nécrose aiguë cornéenne ou kératomalacie. Il existe une corrélation forte entre les signes cliniques de xérophtalmie et la mortalité des enfants atteints [2]. L’Organisation mondiale de la santé (OMS) a mis en place une classification internationale de la xérophtalmie (tableau 9-9) [4].
Tableau 9-9 – Classification internationale de la xérophtalmie selon l’OMS, l’âge de survenue, les risques de cécité et de mortalité associés.
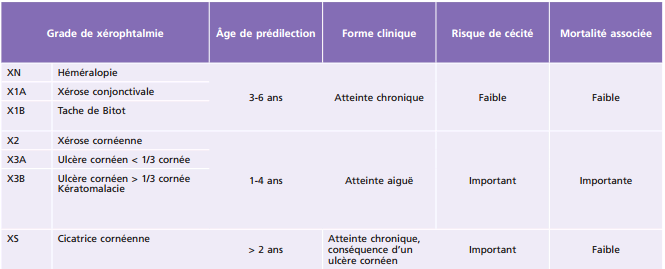
Deux formes cliniques distinctes sont observées :
- la carence chronique en vitamine A : elle atteint essentiellement les enfants entre 3 et 6 ans et est non cécitante. Cette forme est corrélée à un risque faible de mortalité [5]. Elle se manifeste par une héméralopie, une xérose conjonctivale et des taches de Bitot. Celles-ci sont pathognomoniques et correspondent à une lésion en relief, blanche et brillante avec un aspect mousseux sur la conjonctive bulbaire nasale ou temporale, près du limbe. Les taches de Bitot ne disparaissent pas après traitement de la carence en vitamine A ;
- la carence aiguë en vitamine A : il s’agit d’une carence chronique aggravée de façon aiguë par une infection, une diarrhée ou une rougeole intercurrente. Elle atteint essentiellement les enfants entre 1 et 4 ans et est potentiellement cécitante. Elle est corrélée à une surmortalité élevée [5]. Elle se manifeste par la survenue d’un ulcère cornéen dont le risque de surinfection et de kératite infectieuse est important. Cette ulcération peut évoluer vers la kératomalacie (ulcère cornéen atteignant plus de 1/3 de la surface cornéenne) avec un risque majeur de perforation et de nécrose cornéenne en quelques jours. Les complications de cette atteinte font toute la gravité de la maladie avec un risque de cicatrice cornéenne définitive, d’ectasie cornéenne ou de phtyse [5].
Les atteintes extra-oculaires sont caractérisées par :
- des signes cutanés : il existe une atrophie des glandes sébacées et sudoripares qui entraîne un dessèchement cutané et une hyperkératose prédominante à la face externe des membres inférieurs. Une alopécie peut également être associée à une carence en vitamine A [5] ;
- des signes systémiques : la carence en vitamine A entraîne un déficit immunitaire et notamment un déficit de l’immunité cellulaire et un déficit dans la sécrétion d’anticorps ; elle s’accompagne d’une augmentation de la mortalité des pathologies infectieuses notamment pour les infections respiratoires, le paludisme, la rougeole et les diarrhées [5]. Le retard de croissance est un autre effet de la carence en vitamine A, décrit dans la littérature chez l’animal seulement. Il pourrait induire des neuropathies optiques compressives secondaires à une ostéopénie avec hyperostose des os de l’orbite et rétrécissement du canal optique chez l’enfant [6].
Devant un cas clinique d’avitaminose A, il faut mettre en place une enquête alimentaire, complétée par l’usage de marqueurs biologique (rétinol plasmatique < 10 μg/100 ml [3]) ou cytologique (test d’impression conjonctival) afin de mettre en oeuvre immédiatement des mesures curatives et préventives. Cependant, le dosage du rétinol plasmatique est d’interprétation difficile car la concentration du rétinol plasmatique est finement régulée et ne sera abaissée qu’au stade d’épuisement des réserves. Le rétinol est étroitement lié à la concentration sanguine en RBP et en préalbumine [3]. La carence en vitamine A est définie par un taux de RPB inférieur à 0,028 g/l.
La prise en charge de l’avitaminose A nécessite d’envisager un traitement curatif et surtout prophylactique en zones d’endémie.
Sur le plan préventif, il faut lutter contre les conséquences du déficit vitaminique chez les enfants d’âge préscolaire : infections (fièvres éruptives, infections respiratoires), malnutrition, diarrhées. À côté de la morbidité d’origine infectieuse chez l’enfant – rougeole (létalité multipliée par trois), paludisme, diarrhées, infections respiratoires –, il faut citer le rôle du déficit en vitamine A dans la transmission mère-enfant du virus de l’immunodéficience humaine (VIH) et l’effet positif de la vitamine A sur le bilan du fer chez l’enfant et la femme enceinte.
Le traitement curatif de l’avitaminose A consiste en l’administration de palmitate de rétinol à tous les enfants atteints de xérophtalmie. Supplémentation en vitamine A ; palmitate de rétinol par voie orale :
- 50 000 UI chez l’enfant de moins de 6 mois ;
- 100 000 UI entre 6 et 12 mois ;
- 200 000 UI au-dessus de 12 mois.
La supplémentation doit être renouvelée le lendemain et 2 à 4 semaines plus tard.
En cas de malabsorption, on utilise la voie intramusculaire : 50 000 UI/mois de rétinyl palmitate (tableau 9-10) [1].
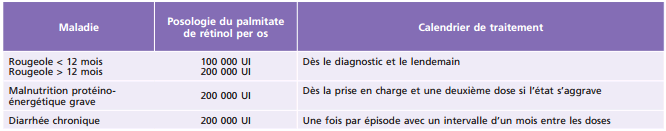
[1] Akrour-Aissou C, Dupré T, Boukari R, et al. Carence en vitamine A chez un groupe d’enfants sains âgés de 1 à 23 mois de la région de Blida, Algérie. Nutrition Clinique et Métabolisme 2014 ; 28 : 4-11.
[2] Sommer A, Tarwotjo I, Hussaini G, Susanto D. Increased mortality in children with mild vitamin A deficiency. Lancet Lond Engl 1983 ; 2 : 585-8.
[3] Amédée-Manesme O, Furr H, Olson JA. Metabolism and function of vitamin A. Arch Fr Pédiatrie 1985 ; 42 : 325-31.
[4] Sommer A. Xerophthalmia and vitamin A status. Prog Retin Eye Res 1998 ; 17 : 9-31.
[5] Gilbert C. The eye signs of vitamin A deficiency. Community Eye Health 2013 ; 26 : 66-7.
[6] Zayed MG, Hickman SJ, Batty R, et al. Unilateral compressive optic neuropathy due to skull hyperostosis secondary to nutritional vitamin A deficiency. Clin Cases Miner Bone Metab 2015 ; 12 : 75-7.
- Chapitre 9
Pathologie de la cornée- 1 – Dystrophies cornéennes héréditaires chez l’enfant
- 2 – Kératocône de l’enfant et de l’adolescent
- 3 – Kératites infectieuses chez l’enfant
- 4 – Kératites allergiques
- 5 – Atteintes cornéennes d’origine systémique
- 6 – Syndromes de Stevens-Johnson et de Lyell chez l’enfant
- 7 – Autres atteintes : kératopathies toxiques chez l’enfant
- 8 – Carence en vitamine A chez l’enfant