Pathologie tumorale intra-oculaire
Coordonné par C. Levy-Gabriel
C. Levy-Gabriel
Le nævus choroïdien est la tumeur intra-oculaire la plus fréquente. Sa prévalence, évaluée dans la littérature uniquement chez l’adulte, varie de 1 à 30 % selon la méthode de dépistage utilisée. Sur le plan anatomopathologique, le nævus correspond à une prolifération mélanocytaire choroïdienne sans signe histologique de malignité (fig. 20-1). Les cellules sont fusiformes, ovoïdes ou rondes, avec un degré variable de pigmentation cytoplasmique. Ces cellules næviques sont probablement présentes dès la naissance mais ne sont visibles que lorsqu’elles se pigmentent progressivement à partir de la puberté. Les nævi choroïdiens sont donc rarement observés chez les jeunes enfants.
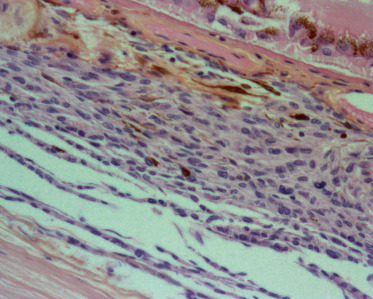
Fig. 20-1 Coupe anatomopathologique d’un nævus choroïdien : prolifération mélanocytaire choroïdienne sans signe histologique de malignité.
Le diagnostic se fait sur l’aspect de la lésion au fond d’oeil, en angiographie (fluorescéinique et au vert d’indocyanine), et en échographie B. Au fond d’oeil, le nævus est une lésion choroïdienne arrondie ou ovalaire à bords flous (fig. 20-2a), de pigmentation variable (souvent peu ou pas pigmentée chez l’enfant). Cette lésion est plane ou très discrètement en relief (fig. 20-2b et 20-3c). La présence de drusen ou d’altérations de l’épithélium pigmentaire (EP) est possible mais moins fréquente chez l’enfant que chez l’adulte (fig. 20-3a). En angiographie fluorescéinique, on note en général une hyperfluorescence irrégulière (fig. 20-3b), variable en fonction du degré de pigmentation du nævus. L’hyperfluorescence est plus marquée lorsque le nævus est achrome, moins détectable si le nævus est déjà très pigmenté (fig. 20-4). En angiographie au vert d’indocyanine, le nævus est hypofluorescent sur les séquences artérielles et veineuses, sans anomalie vasculaire péritumorale (absence de dilatation artérielle ou veineuse, pas de trajectoire vasculaire déplacée). Sur les séquences tardives, l’aspect varie en fonction de l’existence d’altérations de l’EP ou de la rétine. En l’absence de celles-ci, on note une discrète hyperfluorescence.
Les lésions les plus fréquemment confondues avec le nævus sont :
– l’éphélide de la choroïde : lésion pigmentée à bords irréguliers correspondant à une simple augmentation localisée de la pigmentation choroïdienne, qui se différencie du nævus par la présence de gros vaisseaux choroïdiens bien visibles au sein de la lésion ;
– l’éphélide de la choroïde : lésion pigmentée à bords irréguliers correspondant à une simple augmentation localisée de la pigmentation choroïdienne, qui se différencie du nævus par la présence de gros vaisseaux choroïdiens bien visibles au sein de la lésion ;
- le mélanocytome (fig. 20-5) : tumeur localisée le plus souvent au niveau de la papille, plane ou plus souvent légèrement en relief, de coloration uniforme très foncée avec des bords striés et filamenteux.
En pratique, on distingue deux types de tableau clinique (tableau 20-1) :
- le nævus choroïdien bénin typique où l’attitude pratique sera une surveillance régulière ;
- le nævus choroïdien suspect où se pose le problème du diagnostic différentiel avec un petit mélanome de la choroïde.
Le nævus choroïdien est considéré comme d’aspect bénin typique lorsqu’il est découvert fortuitement chez un patient asymptomatique, de petite taille (diamètre < 6 mm et épaisseur < 1,5 mm en échographie) [1], sans pigment orange, sans décollement séreux rétinien (DSR), et sans pin points en angiographie (voir figures fig. 20-2, fig. 20-3b et fig. 20-4c et d). Ce nævus bénin reste stable dans la grande majorité des cas et ne nécessite qu’une surveillance régulière annuelle du fond d’oeil avec rétinophotographies, tomographie par cohérence optique (optical coherence tomography [OCT]), et échographie B pour mesurer l’épaisseur tumorale. Les clichés en lumière rouge sont parfois utiles pour mieux visualiser les limites de la pigmentation. Les complications à type de DSR, néovaisseaux sousrétiniens, oedème rétinien sont très rarement constatées chez l’enfant. Un élargissement discret et très lent du diamètre du nævus peut parfois être constaté sur les rétinophotographies, sans pour autant que cela corresponde à une transformation maligne du nævus. Pour Mashayekhi, cette modification des dimensions concerne 31 % des authentiques nævi choroïdiens à long terme [2].
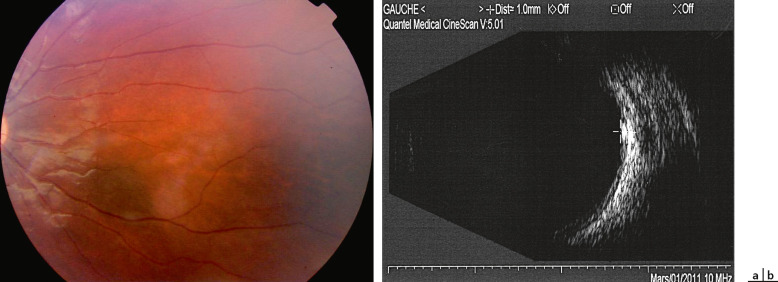
Fig. 20-2 Petit nævus choroïdien bénin typique chez une fillette de 11 ans.
a. Aspect en rétinophotographie : petit diamètre de 2 mm, pas de drusen. b. Aspect échographique avec épaisseur à 1 mm.
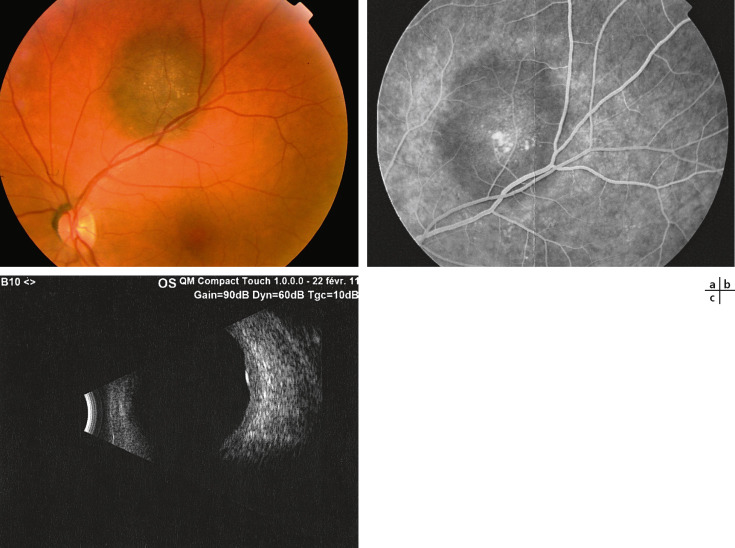
Fig. 20-3 Nævus choroïdien d’aspect typiquement bénin localisé en susmaculaire.
a. Présence de drusen en rétinophotographie. b. Aspect angiographique avec imprégnation des drusen. c. Aspect échographique avec épaississement choroïdien localisé, discrètement hypo-échogène de petit diamètre (4,5 mm) et épaisseur inférieure à 1,5 mm.
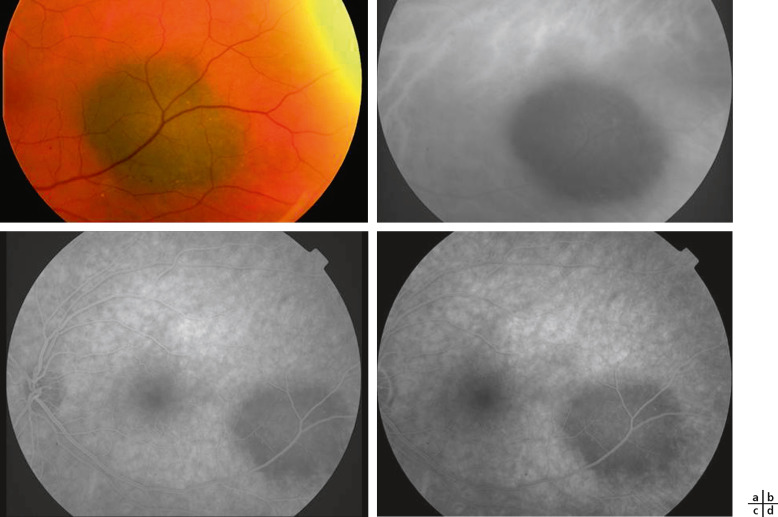
Fig. 20-4 Nævus choroïdien pigmenté.
a. Rétinophotographie. b. Cliché rouge mettant en évidence la pigmentation. c, d. Angiographie en fluorescence : aspect hypofluorescent sur toute la séquence angiographique du nævus.
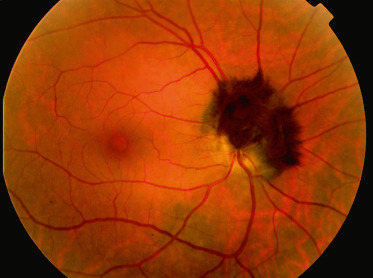
Fig. 20-5 Mélanocytome juxtapapillaire.
À l’opposé du nævus bénin, le nævus suspect se définit par la présence de facteurs de risque de croissance tumorale : symptômes visuels (phosphènes, myodésopsies, baisse d’acuité visuelle) ; pigment orange au fond d’oeil (fig. 20-6 et 20-7) ; dimensions tumorales plus importantes (épaisseur > 1,5 ou 2 mm selon les auteurs, et/ou diamètre > 7 mm) ; localisation à proximité du nerf optique ; présence de pin points en angiographie (hyperfluorescence punctiforme qui augmente discrètement sur les temps tardifs) ; DSR au niveau ou au voisinage immédiat du nævus visible parfois uniquement sur l’OCT [3,4].
En cas de nævus suspect chez un sujet jeune, on optera, en fonction du nombre de facteurs de risque de croissance tumorale, soit pour une surveillance rapprochée avec des contrôles tous les 2 à 3 mois, soit pour un traitement précoce, en mettant en balance les risques de laisser évoluer un petit mélanome et les séquelles attendues du traitement sur la fonction visuelle. En cas de surveillance, celle-ci devra être prolongée car une croissance tumorale peut parfois être observée tardivement, 5 ans voire pour certains plus de 10 ans après l’examen initial. L’intérêt de l’imagerie en autofluorescence dans le cadre de cette surveillance a fait récemment l’objet de quelques publications. Cette technique permet de mettre en évidence les dysfonctionnements de l’EP en particulier ceux se traduisant par l’accumulation excessive de lipofuscine. La présence de drusen, de pigment orange ou d’un DSR au niveau du nævus se manifeste généralement par une hyper-autofluorescence ; à l’opposé, les zones d’atrophie, d’hypertrophie ou de métaplasie fibreuse de l’EP ont tendance à diminuer l’autofluorescence. En cas de petite lésion pigmentée choroïdienne suspecte, cette nouvelle imagerie non invasive apporte donc quelques éléments supplémentaires mais n’a pas permis d’identifier d’argument formel permettant de différencier avec une parfaite fiabilité un nævus bénin d’un petit mélanome débutant [5].
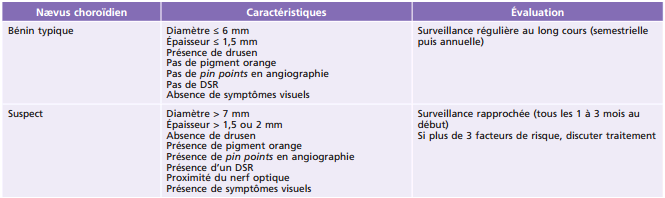
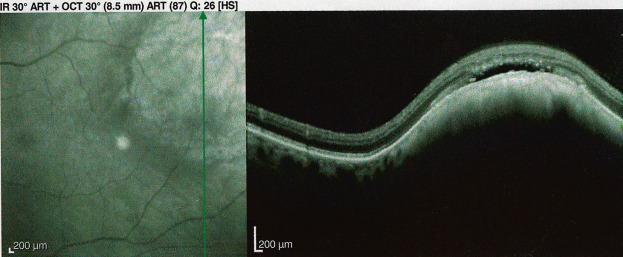
Fig. 20-6 Nævus avec petit DSR en OCT.
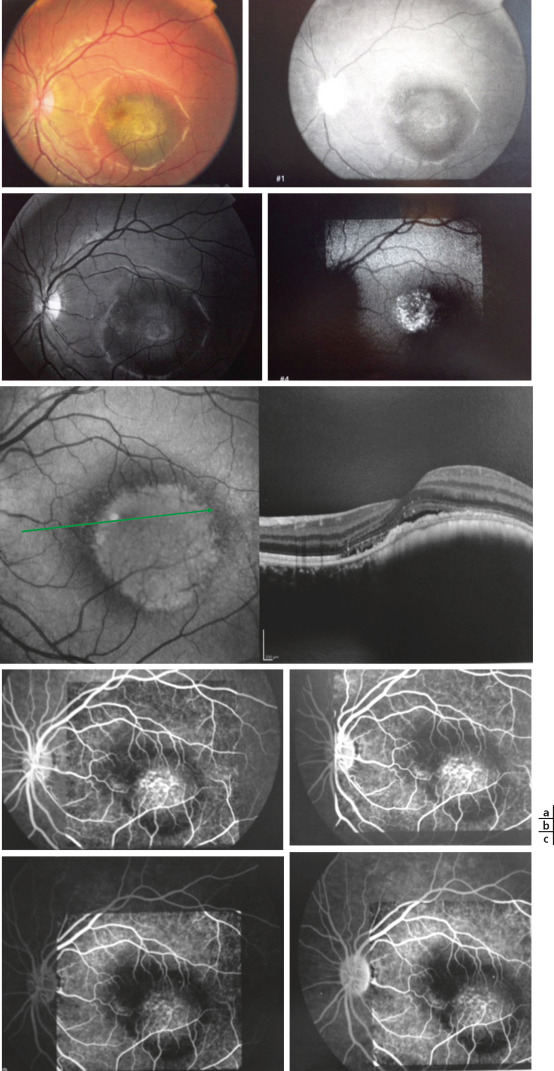
Fig. 20-7 Nævus suspect chez une jeune fille de 13 ans.
a. Rétinophotographie (pigment orange, épaisseur limite à 1,8 mm, diamètre à 5 mm), cliché rouge et en autofluorescence (hyper-autofluorescence des dépôts de lipofuscine). b. Petit DSR en OCT. c. Présence de nombreux pin points en angiographie.
[1] Shields CL, et al. Clinical spectrum of choroidal nevi based on age at presentation in 3422 consecutive eyes. Ophthalmology 2008 ; 115 : 546-552.e2.
[2] Mashayekhi A, et al. Slow enlargement of choroidal nevi : a long-term follow-up study. Ophthalmology 2011 ; 118 : 382-8.
[3] Desjardins L, et al. Risk factors for the degeneration of the choroid naevi : a retrospective study of 135 cases. J Fr Ophtalmol 2001 ; 24 : 610-6.
[4] Shields CL, et al. Choroidal nevus transformation into melanoma: analysis of 2514 consecutive cases. Arch Ophthalmol 2009 ; 127 : 981-7.
[5] Gunduz K, et al. Review of fundus autofluorescence in choroidal melanocytic lesions. Eye (Lond) 2009 ; 23 : 497-503.
C. Levy-Gabriel
L’hypertrophie congénitale de l’EP est une lésion bénigne, congénitale, en général asymptomatique. Elle est constituée en histologie de cellules de l’épithélium pigmenté rétinien hypertrophiques et hyperpigmentées, plus rarement hyperplasiques. Cette tumeur est en général unique et isolée, mais elle peut dans certains cas se présenter sous une forme multiple (aussi appelée pigmentation congénitale groupée de la rétine ou pigmentation en trace d’animaux). La polypose adénomateuse familiale a été associée à une forme particulière d’atteinte multifocale. Au fond d’oeil, l’hypertrophie congénitale de l’EP isolée se présente sous la forme d’une tâche nettement pigmentée, plus ou moins uniforme du fait de la présence de fréquentes lacunes à l’emportepièce (fig. 20-8). L’aspect au fond d’oeil est en général typique, très différent du nævus, du mélanome et du mélanocytome, car totalement plan avec des bords bien nets parfois festonnés [1,2]. En OCT, la rétine neurosensorielle est amincie en regard de la lésion, l’EP est épaissi dans les zones pigmentées et aminci dans les zones lacunaires. Une augmentation de la lésion en surface est souvent constatée au cours du suivi, ainsi qu’une modification de la pigmentation [3]. La survenue d’anomalies vasculaires rétiniennes (raréfaction capillaire, microanévrismes, anastomose choriorétinienne), de membranes néovasculaires ou de rares transformations malignes en adénocarcinome a été décrite [4,5] et justifie une surveillance régulière.
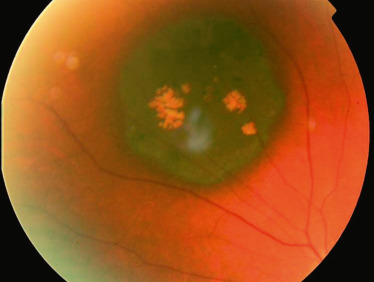
Fig. 20-8 Hypertrophie congénitale de l’épithélium pigmentaire unique isolée.
Dans le cas des pigmentations congénitales groupées, les taches sont plutôt de petite taille, avec une pigmentation relativement homogène et sans lacune, elles sont disposées en amas sur un ou deux secteurs rétiniens. L’atteinte peut être bilatérale. Les lésions sont stables et n’augmentent pas en surface comme les lésions isolées.
La polypose adénomateuse familiale est une affection génétique autosomale dominante résultant d’une mutation constitutionnelle au niveau du gène APC sur le bras long du chromosome 5. Elle se caractérise par l’existence de multiples polypes adénomateux coliques évoluant inéluctablement vers la malignité avant la quarantaine. La prise en charge consiste en une résection chirurgicale prophylactique du côlon et du rectum. Des manifestations extracoliques peuvent être associées : ostéomes, anomalies dentaires, kystes épidermoïdes, tumeurs desmoïdes, et cancers extracoliques thyroïdien, hépatique ou du système nerveux central. La présence au fond d’oeil d’hypertrophie congénitale de l’EP rétinien est la manifestation extra-intestinale la plus fréquente et la plus précocement visible chez l’enfant. Elle est présente dans 90 % des patients porteurs de la mutation APC [6]. Les zones d’hypertrophie congénitale de l’EP peuvent alors être uniques ou multiples, uni- ou bilatérales. Elles mesurent entre 1 et 2 mm. En cas de population à risque, la recherche de ces lésions au fond d’oeil représente donc une méthode fiable, simple et non invasive de dépistage précoce de la polypose adénomateuse.
[1] Fung AT, Pellegrini M, Shields CL. Congenital hypertrophy of the retinal pigment epithelium : enhanced-depth imaging optical coherence tomography in 18 cases. Ophthalmology 1016 ; 121 : 251-6.
[2] Zografos L. Tumeurs et pseudo-tumeurs de l’éputhélium pigmenté et non pigmenté. In : Rapport SFO 2002, Tumeurs intra-oculaires. Paris : Masson ; 2002, chapitre 11, p. 413-61.
[3] Shields CL, et al. Solitary congenital hypertrophy of the retinal pigment epithelium : clinical features and frequency of enlargement in 330 patients. Ophthalmology 2003 ; 110 : 1968-76.
[4] Youhnovska P, Toffoli D, Gauthier D. Congenital hypertrophy of the retinal pigment epithelium complicated by a choroidal neovascular membrane. Digit J Ophthalmol 2013 ; 19 : 24-7.
[5] Cohen SY, et al. Retinal vascular changes in congenital hypertrophy of the retinal pigment epithelium. Ophthalmology 1993 ; 100 : 471-4.
[6] Nusliha A, et al. Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP) ; a polyposis registry experience. BMC Res Notes 2014 ; 7 : 734.
C. Levy-Gabriel
L’hamartome combiné de l’EP et de la rétine est une tumeur bénigne et rare de la rétine neurosensorielle et de l’EP. La plupart des auteurs s’accordent sur l’origine malformative et congénitale de cette tumeur. Elle est en général isolée et peu évolutive, mais peut aussi entrer dans le cadre d’une neurofibromatose de type II dont elle fait partie des critères diagnostics. On retrouve aussi dans la littérature des cas isolés d’association à une neurofibromatose de type I, un syndrome de Gorlin- Goltz, une anomalie Poland, un syndrome oculo-brachio-facial, un angiofibrome juvénile nasopharyngé. Enfin, beaucoup plus rarement, l’hamartome combiné est acquis en rapport avec une réaction hyperplasique de l’EP. Le diagnostic est en général fait dans l’enfance, sur une baisse d’acuité visuelle (40 % ) ou un strabisme (28 % ). On note une prédominance masculine. L’hamartome combiné se présente comme une masse de la rétine neurosensorielle et de l’EP discrètement en relief, plus ou moins pigmentée, brune ou grise, avec une membrane prérétinienne et une traction vitréorétinienne plus ou moins marquée, des vaisseaux rétiniens anormaux, déplacés et tortueux (fig. 20-9). Une exsudation, plus ou moins importante, avec DSR ou des microhémorragies peuvent entourer la lésion. L’atteinte est en général unilatérale, de localisation juxtapapillaire ou papillaire.
La localisation maculaire représente 20 à 40 % des cas, les localisations périphériques sont beaucoup plus rares. Différentes complications évolutives ont été décrites dans la littérature et justifient un suivi à long terme : croissance tumorale, progression de la traction vitréorétinienne avec distorsion fovéolaire, DSR, néovaisseaux prérétiniens, hémorragie intravitréenne [1]. Il n’existe actuellement aucun consensus concernant la prise en charge de ces complications. Pour certains, la réalisation d’une vitrectomie par la pars plana avec ou sans pelage de membrane peut apporter une amélioration de l’acuité visuelle dans 60 % des cas [2]. Pour d’autres, la photocoagulation au laser et les injections intravitréennes de triamcinolone ont aussi un rôle à jouer [3].
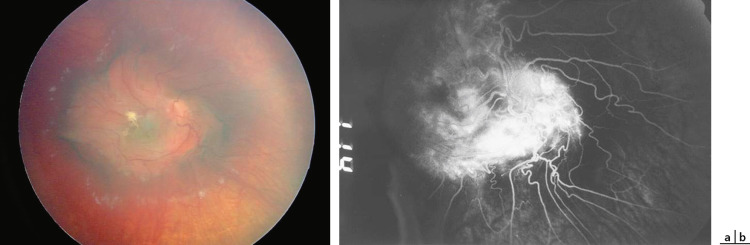
Fig. 20-9 Hamartome combiné de l’EP et de la rétine.
a. Rétinophotographie. b. Aspect angiographique.
[1] Shields CL, et al. Combined hamartoma of the retina and retinal pigment epithelium in 77 consecutive patients visual outcome based on macular versus extramacular tumor location. Ophthalmology 2008 ; 115 : 2246-2252.e3.
[2] Zhang X, et al. Surgical management of epiretinal membrane in combined hamartomas of the retina and retinal pigment epithelium. Retina 2010 ; 30 : 305-9.
[3] Nam DH, et al. Vitrectomy, laser photocoagulation, and intravitreal triamcinolone for combined hamartoma of the retina and retinal pigment epithelium. Ophthalmic Surg Lasers Imaging 2010 ; 9 : 1-4.
C. Levy-Gabriel
L’ostéome choroïdien est une tumeur osseuse bénigne rare, diagnostiquée en général chez les jeunes filles (4 cas sur 5) dans la 2e décennie. L’atteinte est unilatérale dans deux tiers des cas. Au fond d’oeil, on retrouve une masse choroïdienne achrome blanc jaunâtre à bords géographiques bien définis, discrètement en relief, située en général à proximité de la papille (fig. 20-10a). L’aspect calcifié est caractéristique en échographie ou au scanner (fig. 20-10b). Un suivi régulier et à long terme avec rétinophotographies et OCT est recommandé. Une modification de l’ostéome est souvent constatée : augmentation du volume tumoral (40 % des cas pour Shields ; fig. 20-11) ou à l’opposé réduction avec décalcification [1]. Les complications classiquement décrites sont une atrophie progressive de la rétine ou l’apparition d’un DSR en regard de l’ostéome, la présence d’hémorragies rétiniennes avec ou sans néovaisseau choroïdien. Les traitements des néo-vaisseaux rapportés dans la littérature sont la photocoagulation, la photothérapie dynamique, la thermothérapie transpupillaire, la protonthérapie et les injections intravitréennes d’anti-vascular endothelial growth factor (anti-VEGF) [2]. L’acuité visuelle à long terme est souvent très médiocre avec une acuité visuelle inférieure à 1/10 dans 56 % des cas après 10 ans de suivi pour Shields [1].
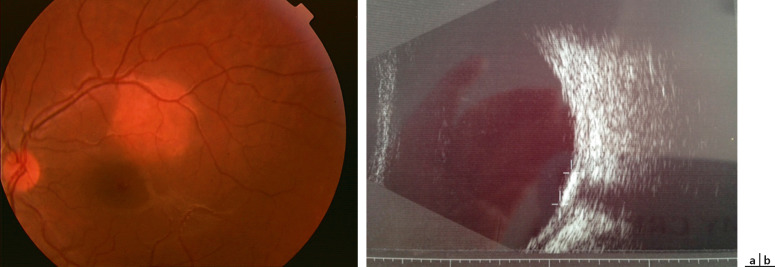
Fig. 20-10 Ostéome choroïdien chez une jeune fille de 15 ans (acuité visuelle : 10/10).
a. Rétinophotographie. b. Aspect plan et calcifié en échographie.
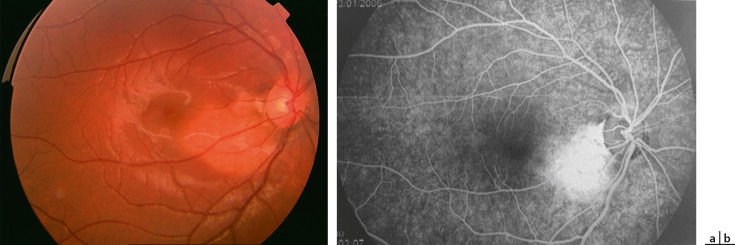
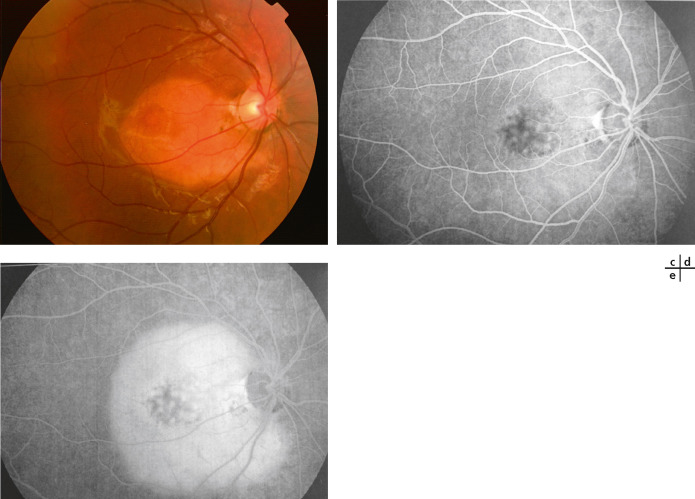
Fig. 20-11 Croissance d’un ostéome choroïdien sur 3 ans.
a. Ostéome en 2004 chez une jeune fille de 13 ans (acuité visuelle : 10/10). b. Aspect angiographique en 2004. c. Augmentation de l’ostéome en diamètre en 2007 : rétinophotographie (acuité visuelle : 10/10). d, e. Aspect angiographique en 2007 à 20 secondes et 4,20 minutes ne notant ni diffusion ni néovaisseaux.
[1] Shields CL, et al. Factors predictive of tumor growth, tumor decalcification, choroidal neovascularization, and visual outcome in 74 eyes with choroidal osteoma. Arch Ophthalmol 2005 ; 123 : 1658-66.
[2] Mansour AM, et al. Role of intravitreal antivascular endothelial growth factor injections for choroidal neovascularization due to choroidal osteoma. J Ophthalmol 2014 ; 2014 : 210458.
C. Levy-Gabriel
Le mélanome uvéal est une tumeur mélanocytaire maligne qui se développe au niveau de la choroïde, du corps ciliaire ou de l’iris. Les cas pédiatriques sont rares et ne représentent que 0,8 à 1,1 % de l’ensemble des mélanomes uvéaux, soit une incidence mondiale annuelle d’environ 65 cas/an. Plus la tranche d’âge est élevée, plus l’incidence augmente, mais la fréquence cumulative de survenue d’un mélanome uvéal n’augmente que de 0,8 % par année d’âge entre 5 et 10 ans, alors qu’elle augmente de 8,8 % par année d’âge chez l’adulte jeune entre 17 et 24 ans [1]. Le développement d’un mélanome uvéal chez l’enfant peut être favorisé par la préexistence d’un nævus d’Ota ou mélanocytose oculodermique : nævus bleu se présentant sous la forme d’une pigmentation bleu-gris de la sclère, congénitale et unilatérale, associée à une mélanocytose irienne (aspect d’hétérochromie irienne avec iris plus foncé présentant des mammillations du côté du nævus) (fig. 20-12) et à une mélanocytose dermique (nævus bleu de la zone cutanée péri-oculaire). Un nævus d’Ota est retrouvé dans 1,9 à 11 % des cas de mélanome uvéal pédiatrique, soit 9 fois plus que dans les cas de mélanomes uvéaux chez l’adulte [2], et 50 à 100 fois plus que dans la population générale. En présence d’un nævus d’Ota, le risque de mélanome uvéal justifie donc la surveillance annuelle du fond d’oeil dès la petite enfance. La neurofibromatose de type I, le B-K mole syndrome, le syndrome des nævi dysplasiques, ainsi que la présence d’une mutation constitutionnelle au niveau du gène BAP1 peuvent aussi favoriser l’apparition d’un mélanome uvéal dans l’enfance. Les caractéristiques cliniques du mélanome au fond d’oeil, en échographie Doppler couleur et en imagerie par résonance magnétique (IRM) (fig. 20-13), ainsi que la prise en charge thérapeutique sont identiques chez l’enfant et chez l’adulte. Les seules différences constatées chez l’enfant sont une proportion plus importante de mélanomes iriens (entre 12 et 39 % [2–4]), une discrète prédominance féminine [1,4,5] et un meilleur pronostic vital avec une survie globale à 92 % à 10 ans [1]. Les facteurs ayant une influence défavorable sur la survie chez l’enfant sont :
- le stade TNM (tumor-node-metastasis) avec une survie sans métastase à 10 ans respectivement de 100 % , 96 % et 82 % pour les catégories T1 (petites tumeurs), T2 (tumeurs moyennes) et T3 (grosses tumeurs) ;
- la génomique tumorale (la monosomie 3 avec addition de 8q exposant au risque métastatique le plus élevé) ;
- le sexe féminin ;
- la préexistence d’un nævus d’OTA (risque de mortalité multiplié par 5–6) [1].

Fig. 20-12 Nævus d’Ota, oeil droit chez un garçon de 7 ans.
a. OEil droit avec mélanocytose uvéale (aspect gris-bleu de la sclère et aspect pigmenté de l’iris). b. OEil gauche. À noter l’hétérochromie irienne.
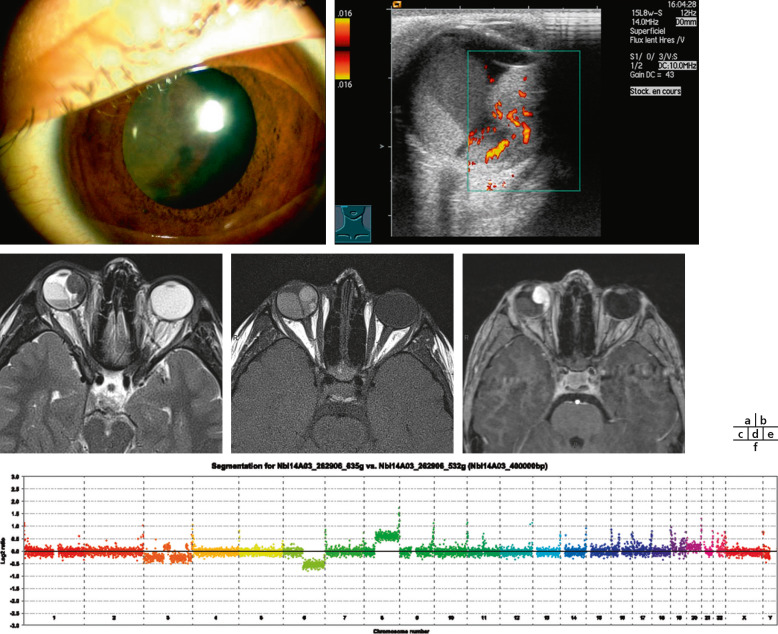
Fig. 20-13 Mélanome ciliochoroïdien chez une fillette de 12 ans.
a. Aspect en lampe à fente. b. Aspect en échographie Doppler couleur. c, d, e. Aspect IRM avec hypersignal en T1, hypersignal en T2 et rehaussement après injection de gadolinium. f. Profil génomique de cette tumeur avec monosomie 3 et addition de 8q (profil à haut risque métastatique).
[1] Al-Jamal RT, et al. The pediatric choroidal and ciliary body melanoma study: a survey by the European Ophthalmic Oncology Group. Ophthalmology 2016 ; 123 : 898-907.
[2] Singh AD, et al. Uveal melanoma in young patients. Arch Ophthalmol 2000 ; 118 : 918-23.
[3] Barr, CC, McLean IW, Zimmerman LE. Uveal melanoma in children and adolescents. Arch Ophthalmol 1981 ; 99 : 2133-6.
[4] Shields CL, et al. Uveal melanoma in teenagers and children. A report of 40 cases. Ophthalmology 1991 ; 98 : 1662-6.
[5] Vavvas D, et al. Posterior uveal melanoma in young patients treated with proton beam therapy. Retina 2010 ; 30 : 1267-71.
C. Levy-Gabriel
Le médullo-épithéliome est une tumeur embryonnaire rare (environ 200 cas publiés dans la littérature), développée à partir de l’épithélium médullaire primitif (partie interne de la cupule optique), le plus souvent au niveau du corps ciliaire (épithélium ciliaire non pigmenté de la pars plicata), beaucoup plus rarement au niveau de l’iris, de la rétine ou de la tête du nerf optique. Sa malignité est variable : selon les séries publiées, entre 20 et 62 % des médullo-épithéliomes sont bénins, entre 38 et 80 % sont malins. Cette tumeur est en général isolée et considérée comme non héréditaire. Mais dans environ 5 % des cas, le médullo-épithéliome apparaît dans le cadre d’un syndrome de prédisposition tumorale avec blastome pleuropulmonaire et mutation constitutionnelle du gène DRCER1 [1,2].
L’atteinte unilatérale se manifeste, en général, dans 75 à 90 % des cas durant la première décennie (moyenne d’âge 5 ans). Les erreurs de diagnostic initial et de prise en charge sont fréquentes (respectivement 88 % et 39 % des cas dans la série de 41 cas publiée par Shields en 2013 [1]). Le tableau classique est celui d’une masse achrome du corps ciliaire, refoulant ou infiltrant l’iris, de couleur variable (grise, blanche, jaune), associée dans 61 % des cas à des kystes intratumoraux bien visibles en échographie. Ces kystes peuvent se détacher de la masse principale et flotter librement dans l’humeur aqueuse ou le vitré (fig. 20-14 et 20-15). Mais la masse tumorale ciliaire est souvent très difficilement visualisable au début et ne se manifeste que par les effets secondaires liés à la croissance tumorale (glaucome secondaire, modifications cristalliniennes, masse pupillaire, extension extra-oculaire). La présence d’une membrane cyclitique néoplasique rétrolentale ou d’une néovascularisation irienne est constatée dans plus de la moitié des cas (51 % ). Enfin, un glaucome (44 % ) ou des anomalies cristalliniennes (cataracte 46 % , subluxation 27 % , colobome cristallinien 20 % ) sont aussi fréquemment associés. En pratique, le médullo-épithéliome est souvent confondu avec un rétinoblastome, une uvéite, plus rarement avec une maladie de Coats ou une persistance du vitré primitif, et le diagnostic n’est parfois fait que sur l’analyse de la pièce d’énucléation.
En anatomopathologie, le médullo-épithéliome est caractérisé par la présence de petites cellules indifférenciées basophiles aunoyau hyperchromatique agencées en cordons anastomotiques ou en rubans, et réalisant parfois des structures kystiques de tailles variées. On peut retrouver des plages d’architecture plus massive constituées de cellules neuroblastiques impossibles à différencier des cellules de rétinoblastome avec des rosettes de Homer- Wright et de Flexner-Wintersteiner. Les médullo-épithéliomes qui contiennent un nombre significatif de cellules indifférenciées avec un index mitotique élevé et un caractère invasif sont considérés comme malins. On distingue les médullo-épithéliomes non tératoïdes, constitués de cellules issues d’une seule couche de tissu embryonnaire germinal et donc uniquement de tissu épithélial, et les médullo-épithéliomes tératoïdes constitués de cellules issues de deux différentes couches de tissu embryonnaire germinal etqui peuvent donc contenir des éléments osseux, cartilagineux ou nerveux.
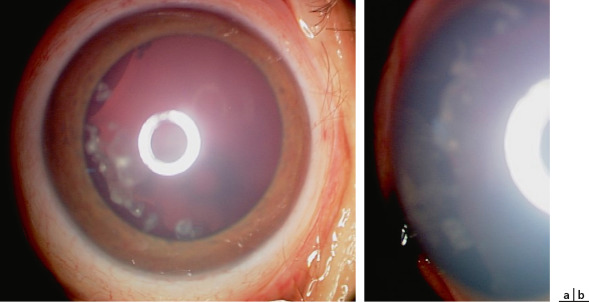
Fig. 20-14 Médullo-épithéliome chez un garçon de 5 ans.
a. Aspect en lampe à fente : noter les kystes tumoraux et les encoches cristalliniennes. La masse ciliaire n’est pas spontanément visible. b. Au verre à trois miroirs, avec indentation, on devine la masse tumorale ciliaire grisâtre.
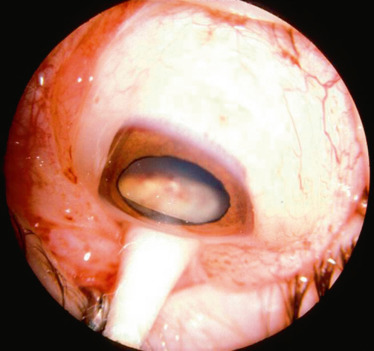
Fig. 20-15 Médullo-épithéliome avec masse ciliaire isolée.
La prise en charge reste mal codifiée. La majorité des cas publiés dans la littérature (60 à 70 % selon les séries) a été traitée par énucléation d’emblée [1, 3,4] et, en cas de tumeur volumineuse, celle-ci reste recommandée par la plupart des auteurs. L’exérèse tumorale par scléro-uvectomie lamellaire partielle ou iridocyclectomie peut être une alternative en cas de petite tumeur limitée à 3 ou 4 quadrants horaires (23 % des cas dans la série de Shields) ; mais en l’absence d’irradiation complémentaire, le risque de récidive locale est très élevé (entre 50 et 100 % [1,3]). Plus récemment, la curiethérapie par disque d’iode 125 ou plaque de ruthénium 106 a été utilisée avec succès, soit en traitement complémentaire après exérèse tumorale localisée, soit en traitement primaire de la tumeur [1,5,6]. Les indications de radiothérapie externe (traitement des récidives, traitement adjuvant en cas d’atteinte extrasclérale ou orbitaire), ainsi que les indications de cryothérapie (traitement des récidives locales minimes) sont beaucoup plus limitées et ne représentent plus chacune que 3 % des cas. Le rôle de la chimiothérapie n’est pas encore très bien établi.
L’évolution après traitement est en général favorable. Dans la série de Shields, le taux de dissémination métastatique est de seulement 8 % et tous les patients métastatiques avaient initialement une tumeur très évoluée avec extension orbitaire.
[1] Kaliki S, et al. Ciliary body medulloepithelioma : analysis of 41 cases. Ophthalmology 2013 ; 120 : 2552-9.
[2] Priest JR, et al. Ciliary body medulloepithelioma : four cases associated with pleuropulmonary blastoma--a report from the International Pleuropulmonary Blastoma Registry. Br J Ophthalmol 2011 ; 95 : 1001-5.
[3] ECanning CR, McCartney AC, Hungerford J. Medulloepithelioma (diktyoma). Br J Ophthalmol 1988 ; 72 : 764-7.
[4] Holdt M, et al. Intraocular medulloepithelioma – series of 10 cases and review of the literature. Klin Monbl Augenheilkd 2009 ; 226 : 1017-22.
[5] Poon DS, et al. Ruthenium-106 plaque brachytherapy in the primary management of ocular medulloepithelioma. Ophthalmology 2015 ; 122 : 1949-51.
[6] Cassoux N, et al. Conservative surgical treatment of medulloepithelioma of the ciliary body. Arch Ophthalmol 2010 ; 128 : 380-1.
C. Levy-Gabriel
Le léiomyome est une tumeur rare, bénigne, développée à partir des fibres musculaires lisses. L’atteinte oculaire est le plus souvent uvéale au niveau du corps ciliaire ou de l’iris (léiomyome mésectodermal) ; beaucoup plus rarement, le léiomyome se développe à partir des fibres musculaires entourant les vaisseaux sanguins intra-oculaires (léiomyome mésodermal) [1,2]. Moins de 70 cas ont été rapportés dans la littérature. On note une légère prédominance féminine. Chez l’enfant, cette tumeur est exceptionnellement diagnostiquée avant l’âge de 16 ans. À l’examen, la tumeur peut être achrome (rosée ou blanc grisâtre) ou pigmentée, elle s’accompagne dans un quart des cas d’un DR exsudatif. Le léiomyome présente souvent une croissance tumorale lente ; un envahissement de la chambre antérieure, une luxation du cristallin, un glaucome ou un envahissement extrascléral ont été décrits. La masse en dôme peut être hypoéchogène ou iso-échogène en échographie. En IRM, elle est hyperintense par rapport au vitré en T1, hypo-intense en T2, et se rehausse après injection de gadolinium. Aucune caractéristique de cette tumeur n’est pathognomonique (fig. 20-16) et son aspect est très difficile à différencier des autres tumeurs intra-oculaires, en particulier du mélanome uvéal [3]. Les arguments qui doivent faire évoquer un léiomyome sont le terrain (patient jeune, de sexe féminin, mélanoderme), la localisation tumorale au corps ciliaire surtout lorsqu’en échographie elle est supra-uvéale, le caractère transilluminable de la lésion. Le diagnostic est cependant souvent uniquement histologique retrouvant une tumeur d’origine myogénique avec en immunohistochimie un marquage positif de l’actine, pas d’expression de la protéine S100, ni de l’HMB45. Le traitement dépend du volume tumoral et de la localisation. Pour les plus petites tumeurs, il est possible de proposer une surveillance, mais en pratique c’est rarement le cas dans la mesure où le diagnostic de mélanome ne peut pas être formellement éliminé. Si la tumeur iridociliaire intéresse moins de 3 à 4 quadrants horaires, une exérèse chirurgicale par iridectomie ou scléro-uvectomie lamellaire partielle peut être réalisée [4,5]. Un cas de récidive locale après exérèse a cependant été rapporté. Les tumeurs les plus volumineuses, en général confondues avec un mélanome, sont traitées par énucléation ou radiothérapie. Récemment, la présence de récepteurs aux hormones stéroïdes a été mise en évidence (récepteurs à la progestérone dans deux cas de léiomyome chez des femmes ; récepteurs aux androgènes dans un cas survenu chez un homme) ouvrant la porte à d’éventuelles hormonothérapies [6].
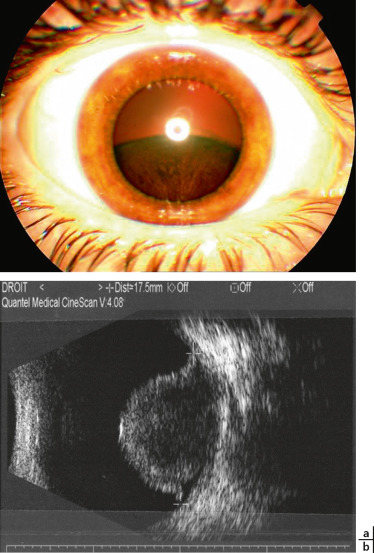
Fig. 20-16 Léiomyome chez un jeune homme de 16 ans.
a. Aspect de masse tumorale ciliaire pigmentée très difficile à différencier d’un mélanome. b. Aspect échographique avec masse hypo-échogène en dôme.
[1] Koletsa T, et al. Mesectodermal leiomyoma of the ciliary body : report of a case and review of the literature. Pathol Res Pract 2009 ; 205 : 125-30.
[2] Shields JA, et al. Observations on seven cases of intraocular leiomyoma. The 1993 Byron Demorest Lecture. Arch Ophthalmol 1994 ; 112 : 521-8.
[3] Remmer MH, et al. Giant leiomyoma of the ciliary body. Oman J Ophthalmol 2014 ; 7 : 81-3.
[4] Razzaq L, et al. Mesectodermal suprauveal iridociliary leiomyoma : transscleral excision without postoperative iris defect. Arch Ophthalmol 2011 ; 129 : 1635-7.
[5] Richter MN, et al. Transscleral resection of a ciliary body leiomyoma in a child : case report and review of the literature. Graefes Arch Clin Exp Ophthalmol 2003 ; 241 : 953-7.
[6] Quhill H, et al. Three cases of intraocular mesectodermal leiomyoma expressing progesterone and androgen receptors. Eye 2013 ; 27 : 669-72.
L. Lumbroso-Le Rouic
Le rétinoblastome est la tumeur intra-oculaire maligne primitive la plus fréquente de l’enfant. Il s’agit pourtant d’une tumeur rare dont l’incidence est d’environ 1/15 000 naissances [1]. Le rétinoblastome est une maladie complexe du fait de sa rareté, de son risque vital mais aussi fonctionnel chez de jeunes enfants, et de la nature génétique de la maladie.
Le rétinoblastome est bilatéral dans 40 % des cas, l’âge médian de découverte est alors inférieur à 1 an et tous les patients sont porteurs d’une mutation constitutionnelle du gène du rétinoblastome (RB1) situé sur le bras long du chromosome 13 (13q14). Dans 60 % des cas, le rétinoblastome est unilatéral, l’âge médian au moment du diagnostic est alors plus élevé (24 mois) et 15 % des patients sont porteurs d’une mutation constitutionnelle de RB1. Le pronostic vital est excellent dans les pays industrialisés, avec une survie de plus de 95 % [2]. Le pronostic oculaire et la fonction visuelle dépendent en revanche de l’étendue de l’atteinte oculaire initiale. La conservation oculaire n’est pas toujours possible dans les formes intra-oculaires étendues.
Les patients porteurs de la mutation du gène RB1 ont une prédisposition aux cancers : le rétinoblastome dans la petite enfance, des sarcomes à l’adolescence et d’autres pathologies malignes à l’âge adulte.
Ces patients ont par ailleurs un risque de transmettre l’anomalie génétique à leur descendance selon un mode autosomique dominant, avec une pénétrance d’environ 90 % . Ceci nécessite de proposer une consultation de génétique à tous les sujets atteints de rétinoblastome afin d’instituer les modalités de surveillance ophtalmologique pour leur descendance ainsi que pour leurs apparentés.
Le diagnostic du rétinoblastome est avant tout clinique. Il s’agit d’une tumeur rétinienne blanche, caractéristique, découverte à l’examen du fond d’oeil (FO) réalisé lors de la constatation d’un signe d’appel.
La grande majorité des enfants atteints de rétinoblastome sont en âge préverbal. Le principal signe d’appel est la constatation par les parents ou les proches d’une leucocorie (fig. 20-17), La constatation ou même la simple mention par des parents de la présence d’une leucocorie doit faire évoquer un rétinoblastome et l’examen du FO doit être réalisé en urgence. Il permet de confirmer la présence d’une ou de plusieurs tumeurs. Le deuxième signe d’appel est le strabisme, qui lui aussi nécessite un avis ophtalmologique rapide avec examen du FO pour éliminer le rétinoblastome, mais aussi d’autres pathologies organiques.
Dans les pays développés, les autres circonstances de découverte sont beaucoup plus rares, cela peut aller du dépistage systématique d’enfants à risque (apparentés à un patient traité pour rétinoblastome) à un tableau inflammatoire ou tumoral orbitaire très « bruyant » avec une buphtalmie.
De façon générale, toute anomalie oculaire (hétérochromie, buphtalmie, etc.) chez un petit enfant doit faire pratiquer un FO et, si celui-ci n’est pas accessible, une imagerie oculaire avec échographie voire IRM pour éliminer une tumeur.
Il s’agit d’une présentation clinique dont le tableau peut faire évoquer une pathologie inflammatoire avec présence d’un hypopion et de flocons vitréens, sans masse tumorale véritable identifiable au FO. Cette forme particulière est un véritable piège diagnostique, d’autant plus qu’elle survient chez des enfants généralement plus âgés. L’absence des autres signes inflammatoires classiques (hyperhémie conjonctivale, synéchies, etc.) doit faire évoquer le diagnostic qui peut être conforté par la présence d’une rétine souvent décollée et épaissie.
Un dépistage systématique doit être effectué chez les enfants à risque de rétinoblastome par la réalisation d’un FO en dehors de tout signe d’appel. Cette surveillance est proposée aux apparentés d’une personne ayant eu un rétinoblastome. Astreignante, elle permet de dépister des tumeurs débutantes et améliore nettement la prise en charge et la conservation oculaire [3]. Les enfants porteurs d’une anomalie du gène RB1 ou ayant un parent atteint de rétinoblastome bilatéral nécessitent un FO dès la première semaine de vie puis tous les mois. Si l’anomalie du gène RB1 n’est pas retrouvée dans la famille, les modalités et la fréquence des contrôles sont adaptées aux risques théoriques de développer la maladie en fonction du degré de parenté.
Ce dépistage doit aussi être réalisé dans le cadre du suivi d’enfants porteurs d’anomalies chromosomiques impliquant le chromosome 13 (syndrome associant un retard psychomoteur, et de croissance), en effet le gène RB1 étant localisé sur ce même chromosome, ces enfants sont à risque de développer un rétinoblastome en plus des autres anomalies.

Fig. 20-17 Leucocorie de l’oeil droit constatée par les parents sur une photographie.
Lorsque le FO fait devant un signe d’appel a montré une masse compatible avec un rétinoblastome, la prise en charge se poursuit en urgence et en milieu spécialisé.
L’examen du fond d’oeil sous anesthésie générale constitue la première étape de cette prise en charge. Il s’agit d’une confirmation diagnostique mais surtout de l’évaluation tumorale initiale de la maladie, ce qui permet d’en effectuer sa classification et de proposer un traitement.
L’aspect des tumeurs peut être variable mais la plupart du temps, il est caractéristique et le diagnostic de rétinoblastome est clinique. Il s’agit de lésions tumorales rétiniennes blanches dont le nombre est variable et pouvant contenir des calcifications (fig. 20-18). La masse tumorale peut être plus ou moins importante : lorsque la masse est très volumineuse, elle est souvent associée à un DR exsudatif. Dans le cas des dépistages systématiques des enfants à risque, des lésions parfois millimétriques sont détectées (fig. 20-19). Un essaimage tumoral sous-rétinien (fig. 20-20a) ou intravitréen (fig. 20-20b), dont la présence et l’étendue sont un facteur de moins bon pronostic de conservation oculaire, peut s’associer à la masse tumorale.
Dans quelques très rares cas, des tumeurs peuvent apparaître, se développer et ne plus évoluer. Il s’agit de « rétinocytomes » ou de rétinoblastomes dits « spontanément involués » ou « spontanément régressifs ». Cliniquement, l’aspect est comparable à celui d’une tumeur cicatricielle, mais survenant chez un patient n’ayant reçu aucun traitement et la plupart du temps asymptomatique (fig. 20-21). Cet aspect est parfois retrouvé au FO chez les parents d’un enfant pris en charge pour rétinoblastome, ce qui signe son caractère héréditaire et familial.
Cette présentation clinique est très rare (moins de 2 % des cas de rétinoblastome). Cliniquement, l’aspect est comparable à une lésion traitée avec une masse tumorale translucide associée à des zones calcifiées et souvent entourée d’altérations réactionnelles de l’épithélium pigmenté rétinien. Une surveillance est recommandée en raison des cas décrits de réévolutivité tumorale. Un bilan avec consultation génétique est aussi nécessaire.
Dans cette présentation clinique aussi très rare (2 % de tous les rétinoblastomes), il n’y a pas de masse tumorale rétinienne classique. Les enfants présentent souvent une atteinte du segment antérieur avec aspect de pseudo-uvéite, parfois des nodules iriens, une néo-vascularisation de l’iris y est parfois associée. Au FO, il peut exister des éléments nodulaires dans le vitré qui peuvent faire évoquer une pathologie inflammatoire. La rétine est souvent infiltrée et blanchâtre, épaissie en échographie ou imagerie oculaire par IRM, mais sans masse tumorale rétinienne classique.
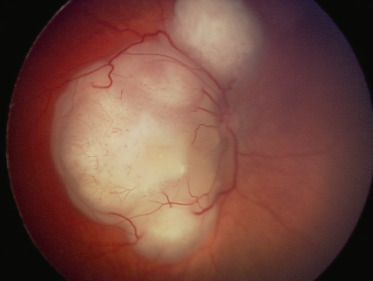
Fig. 20-18 Aspect caractéristique de rétinoblastome avec présence de plusieurs masses tumorales blanches contenant quelques calcifications.
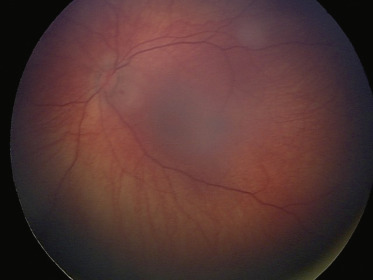
Fig. 20-19 Découverte de deux petites lésions tumorales au FO systématique d’un enfant dont la mère avait été traitée pour rétinoblastome unilatéral.
Le diagnostic différentiel le plus fréquent du rétinoblastome et parfois aussi le plus difficile est une maladie de Coats évoluée [4]. Seul l’examen histologique réalisé en cas de doute diagnostique permettra d’affirmer la bénignité ou pas de la pathologie oculaire. Les autres pathologies sont moins fréquemment confondues avec un rétinoblastome. Il peut s’agir de colobomes, de lésions choriorétiniennes cicatricielles, de DR non tumoraux, d’une persistance du vitré primitif, voire d’autres pathologies tumorales oculaires rares tels l’astrocytome ou le médullo-épithéliome.
Le premier FO d’évaluation permet de réaliser une évaluation tumorale précise en fonction de la taille et de la position des tumeurs. La classification actuellement utilisée, et la plus adaptée aux traitements actuels, est l’International Retinoblastoma Classification (IRC ; tableau 20-2) [5]. Elle permet d’évaluer les chances de conservation oculaire avec les traitements actuellement utilisés, qui ne recourent plus à la radiothérapie externe en première intention, mais le plus souvent à une association de chimiothérapie par voie intraveineuse ou intra-artérielle et des traitements focaux sur chaque site tumoral. La classification de Reese-Ellsworth, adaptée aux traitements par radiothérapie externe conventionnelle n’est guère plus utilisée ou alors qu’à but comparatif pour les publications.
L’atteinte oculaire est classée en fonction de la taille de la tumeur (en cas de tumeurs multiples la plus grande est prise en compte), de sa distance par rapport à la macula et à la tête du nerf optique et de la présence ou pas d’un essaimage dans le vitré ou sous-rétinien (localisé ou à distance de la tumeur).
Le diagnostic du rétinoblastome est le plus souvent clinique. Lorsque le FO n’est pas accessible, une échographie en mode B et une IRM sont réalisées pour conforter le diagnostic. Ils permettent de visualiser une masse tumorale contenant les calcifications caractéristiques.
Le bilan comprend : une IRM orbitaire et cérébrale pour rechercher une atteinte du nerf optique et des pathologies cérébrales exceptionnellement associées au rétinoblastome (pinéaloblastome) ; une prise en charge par un pédiatre oncologue pour un examen clinique. Un bilan général est rarement indiqué d’emblée lorsque les tumeurs sont strictement intra-oculaires et pas trop étendues. Il sera proposé en cas de lésions intra-oculaires très étendues au moment du diagnostic (buphtalmie) ou en cas de constatation en IRM d’une atteinte du nerf optique ou extrasclérale. Ce bilan consiste en une ponction médullaire, une biopsie de moelle et une ponction lombaire.
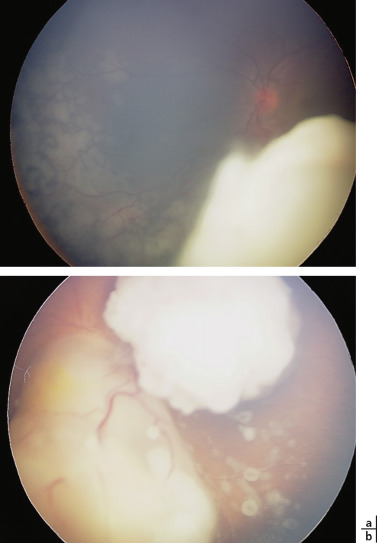
Fig. 20-20 Essaimage tumoral à distance de la tumeur rétinienne.
a. Essaimage sous-rétinien à distance d’un volumineux rétinoblastome. b. Bulles d’essaimage vitréen associées aux tumeurs rétiniennes.
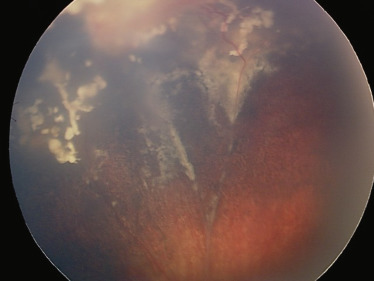
Fig. 20-21 Découverte fortuite d’une lésion du FO avec aspect de rétinoblastome spontanément involué : remaniements de l’EP et aspect fragmenté cicatriciel.

Tableau 20-2 - Classification IRC d’après Murphree [5].
La prise en charge et le suivi sont complexes et multidisciplinaires : ophtalmologistes, pédiatres oncologues, radiologues, anatomopathologistes et généticiens.
Le traitement dépend de multiples paramètres : caractère uniou bilatéral des tumeurs, volume tumoral, localisation par rapport à la macula et/ou le nerf optique, âge de l’enfant et éventuelles pathologies associées.
L’énucléation reste nécessaire lorsque le volume tumoral est très important ne permettant pas un traitement conservateur (tumeur envahissant toute la cavité vitréenne, sans aucun potentiel visuel). L’IRM oculaire est indispensable avant l’intervention pour vérifier l’absence d’extension au niveau du nerf optique ou extrasclérale (fig. 20-22). Si l’imagerie montre une atteinte très étendue (atteinte extrasclérale ou du nerf optique), l’intervention sera précédée d’une chimiothérapie. En effet, il est indispensable que la section du nerf optique soit réalisée en zone saine. L’examen anatomopathologique est un élément capital pour la poursuite de la prise en charge avec réalisation ou pas d’une chimiothérapie complémentaire, voire d’une irradiation orbitaire, en fonction descritères histologiques de risque métastatique ou de récidive orbitaire [6,7]. Lorsque le volume tumoral, le permet un traitement conservateur est proposé.
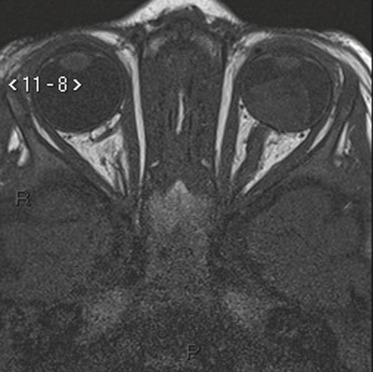
Fig. 20-22 IRM préopératoire avant énucléation d’un rétinoblastome unilatéral évolué : pas d’atteinte du nerf optique.
La stratégie thérapeutique est complexe et souvent plusieurs traitements différents doivent être associés entre eux pour obtenir le contrôle tumoral et la conservation oculaire. Néanmoins, pour de toutes petites lésions, un traitement oculaire seul peut suffire.
La cryoapplication est couramment utilisée depuis de nombreuses années. Il s’agit d’une triple cryoapplication de la tumeur sous contrôle ophtalmoscopique. Cette technique est réalisable pour les plus petites tumeurs (< 3 mm de diamètre), antérieures à l’équateur. Son efficacité est proche de 80 % . Cette technique n’a aucune efficacité sur un essaimage vitréen.
La photocoagulation laser est une technique qui était proposée pour les plus petites tumeurs postérieures à l’équateur mais qui actuellement a été suplantée par la thermothérapie transpupillaire seule ou en association avec une chimiothérapie (thermochimiothérapie).
La thermothérapie transpupillaire (TTT) consiste en une hyperthermie sur chaque site tumoral réalisée à l’aide d’un laser diode, émettant dans les infrarouges. Le but est l’obtention d’une lente élévation de la température dans la tumeur et non une photocoagulation. Les paramètres sont adaptés avec des spots larges (0,8 à 1,2 mm) et des durées d’exposition très longues pouvant aller jusqu’à plusieurs minutes sur chaque site tumoral. Utilisée seule, elle peut être efficace pour les plus petites tumeurs diagnostiquées précocement, en particulier chez des enfants ayant des FO de dépistage systématique.
Pour les lésions de plus de 3 mm, la TTT seule est insuffisante et est alors associée à une chimiothérapie soit intraveineuse soit intra-artérielle pour en potentialiser les effets.
La brachythérapie ou curiethérapie est une irradiation localisée, proposée pour le traitement des tumeurs périphériques non accessibles aux autres traitements (> 3 mm) ou associées à un essaimage vitréen localisé. Le disque est posé sur la sclère en regard de la lésion. Les radio-isotopes les plus fréquemment utilisés sont l’iode 125 ou le ruthénium 106.
La majorité des enfants pris en charge pour rétinoblastome ont des tumeurs dont la taille au diagnostic ne permet pas la réalisation seule de cryoapplication, TTT, voire curiethérapie : une chimiothérapie est nécessaire afin de réduire le volume tumoral et rendre les tumeurs accessibles au traitement local qui sera alors associé au traitement général.
La chimiothérapie peut être réalisée par voie intraveineuse ; les protocoles d’administration, les médicaments utilisés et le nombre de cures sont variables, ils dépendent de l’étendue de la maladie. Pour les atteintes peu évoluées, deux cycles de chimiothérapie sont réalisés : étoposide et carboplatine. Si la réponse est satisfaisante, la chimiothérapie sera continuée par du carboplatine seul et associée à la thermothérapie de chaque site tumoral : on parlera de thermochimiothérapie. L’injection de chimiothérapie est, dans ce cas, suivie très rapidement (1 à 3 heures) par la réalisation sur chaque site tumoral d’une TTT. Il s’agit d’un traitement très efficace sur le contrôle tumoral [8].
Pour les atteintes les plus étendues, une chimiothérapie avec trois médicaments (étoposide, carboplatine et vincristine) est administrée pour 6 cycles avec l’association de traitements sur chaque site tumoral à partir du troisième cycle.
Les résultats des traitements par chimiothérapie intraveineuse et traitements locaux sont satisfaisants avec un contrôle tumoral excellent pour les yeux les moins atteints (groupe A à C). Les yeux ayant une atteinte plus évoluée (groupe D) ont un risque plus important de rechute tumorale et d’énucléation secondaire.
Depuis une dizaine d’année, l’administration de la chimiothérapie peut aussi se faire par injection intra-artérielle sélective directement dans l’artère ophtalmique après cathétérisme fémoral. Le médicament le plus couramment utilisé est le melphalan, mais d’autres molécules telles que le carboplatine ou le topotécan peuvent y être associées.
Un traitement par cryoapplication et TTT sur les tumeurs est régulièrement proposé. Les résultats sont intéressants en termes de contrôle tumoral et conservation oculaire [9].
La tolérance générale est bonne, cependant des effets secondaires oculaires avec risque d’occlusions vasculaires ont été décrits et le pronostic fonctionnel des yeux traités par cette technique reste à évaluer à moyen ou long terme [10,11].
Quelle que soit la stratégie thérapeutique utilisée, une grande partie des échecs tumoraux sont liés à la présence d’un essaimage dans le vitré. Cet essaimage, parfois présent au diagnostic peut aussi apparaître en cours de traitement ou de surveillance. Sa prise en charge est plus difficile, car les traitements focaux sont dans ce cas sans efficacité. La chimiothérapie utilisée par voie veineuse ou intra-artérielle a peu d’effet sur une atteinte vitréenne en raison de la faible pénétration intra-oculaire des médicaments utilisés. Lorsque l’atteinte vitréenne est très localisée et périphérique, elle peut être traitée par une irradiation par disque radio-actif. Mais en cas d’essaimage étendu, ou dans le vitré central, l’irradiation externe a été longtemps la seule possibilité thérapeutique.
C’est dans cette situation clinique que l’utilisation d’une chimiothérapie par voie intravitréenne peut être discutée.
Les injections intravitréennes (IVT) de chimiothérapie représente une technique qui permet une excellente pénétration intra-oculaire d’un médicament. Cependant, dans le cas d’une pathologie tumorale telle que le rétinoblastome, la réalisation de ce geste n’est pas dénuée de risque d’essaimage orbitaire. C’est pour cela que la réalisation récente de chimiothérapie par IVT n’est proposée et réalisée dans cette pathologie que dans des situations cliniques très particulières : atteinte vitréenne isolée, modérée à distance de l’ora serrata, sans lésion rétinienne active associée. Le but est d’éviter une irradiation externe conventionnelle ou une énucléation.
Afin d’éviter un essaimage orbitaire, l’injection doit se faire à distance d’une zone tumorale active. Elle ne doit pas être réalisée si le vitré est envahi de façon massive ou au niveau du site d’injection. Si des tumeurs rétiniennes sont actives, elles doivent être traitées de façon habituelle afin d’éviter un réensemencement. La molécule utilisée est essentiellement le melphalan. Les premiers résultats en matière de contrôle tumoral sont bons, mais le recul est encore très limité. Cette technique paraît intéressante et pourra être utilisée dans des cas très particuliers, cependant sa toxicité et ses risques à moyen ou long terme restent encore à évaluer [12].
Le traitement conservateur est proposé au moins pour un côté dans les formes bilatérales et dans certaines formes unilatérales lorsque le volume tumoral le permet. Le but est d’obtenir le contrôle tumoral en évitant le recours à une énucléation voire l’irradiation externe. La stratégie thérapeutique est décidée au décours du FO d’évaluation initial en milieu spécialisé et adaptée : à l’étendue tumorale, au caractère uni- ou bilatéral de la maladie, à l’âge de l’enfant et à d’éventuelles pathologies associées. Une chimiothérapie intra-artérielle peut être proposée dans les atteintes unilatérales. Une chimiothérapie intraveineuse est souvent proposée pour les atteintes bilatérales. Dans les deux cas, un traitement par TTT ou cryoapplication y est associé, souvent après une ou deux cures de réduction tumorale.
Le pronostic vital d’un enfant atteint de rétinoblastome est excellent dans les pays occidentaux. En effet, la plupart du temps, les enfants ont un bon taux de survie, au prix de la perte de la vision d’un oeil, voire exceptionnellement des deux yeux [2]. Le pronostic est donc essentiellement oculaire et visuel. Le contrôle tumoral peut nécessiter une énucléation, et même en cas de conservation oculaire la présence de localisations tumorales du pôle postérieur peut entraîner une mauvaise vision de façon bilatérale. Néanmoins, la majorité des enfants pris en charge a une acuité visuelle bilatérale satisfaisante de plus de 6/10 [13].
Il faut également souligner que les enfants porteurs de l’anomalie du gène RB1 ont un risque de développer d’autres cancers à l’adolescence ou à l’âge adulte qui peuvent donc entraîner une diminution de leur espérance de vie [14].
Le pronostic vital reste en revanche menacé dans les pays émergents où les décès sont encore fréquents liés à la propagation dans le système nerveux central de la tumeur oculaire évoluée après envahissement du nerf optique. La survie dépend du niveau socioéconomique et varie entre 40 et 80 % des enfants.
À la fin du traitement initial, une surveillance clinique ophtalmologique par FO sous anesthésie générale associée à un suivi pédiatrique est nécessaire, afin de vérifier l’absence de récidive et de dépister l’apparition de nouvelles tumeurs. Ces examens répétés permettent aussi de rechercher les complications liées aux traitements (complications oculaires et générales en cas de traitement par chimiothérapie). Il est également nécessaire de sensibiliser la famille puis l’enfant devenu adolescent et adulte au risque de deuxième tumeur lorsqu’il est porteur d’une anomalie du gène RB1.
L’examen du FO est réalisé sous anesthésie générale de façon mensuelle au décours de la prise en charge initiale, le rythme des contrôles est ensuite espacé progressivement jusqu’à tous les 3 mois. L’anesthésie générale est nécessaire jusqu’à ce que la coopération de l’enfant permette la réalisation d’un FO dans de bonnes conditions.
En l’absence de données génétiques disponibles, un suivi de dépistage des apparentés des enfants atteints est aussi nécessaire. Le schéma de suivi est adapté à la forme présentée par l’enfant (uni- ou bilatérale) et au degré de parenté des enfants de la famille [3]. À l’âge adulte, il faut sensibiliser les patients atteints sur le risque d’être porteurs d’une anomalie génétique transmissible à leur descendance. Une consultation de génétique est systématiquement proposée aux familles d’un enfant traité. Une fois devenu adulte, une nouvelle consultation génétique pourra lui être proposée si elle n’a pas été réalisée auparavant ou pour réinformer le patient.
La consultation génétique est une étape indispensable de la prise en charge d’un patient traité pour rétinoblastome. Elle est actuellement proposée et réalisée assez rapidement au décours de la prise en charge initiale. L’entretien est l’occasion de sou-ligner la possibilité que l’enfant soit porteur d’une anomalie du gène RB1 ainsi que les risques associés. La recherche de cette anomalie constitutionnelle est proposée de façon systématique au décours de la consultation. Une fois cette recherche complétée, le patient et sa famille sont revus et des explications complémentaires sont données en fonction de la détection ou pas d’une anomalie du gène RB1. Ces entretiens sont aussi l’occasion de réaliser un conseil génétique si les parents ont en projet d’avoir d’autres enfants et de l’adapter en fonction de la détection ou pas de l’anomalie génétique. À l’âge adulte, une nouvelle consultation sera proposée à tout patient traité dans l’enfance pour rétinoblastome afin de le sensibiliser à nouveau aux risques pour sa descendance et lui rappeler la nécessité du suivi des enfants à naître en cas de projet parental : un dépistage systématique doit être réalisé pour les enfants à naître de tout parent ayant eu un rétinoblastome dans l’enfance porteur ou non du gène RB1.
Le rétinoblastome est une maladie cancéreuse rare, complexe, génétique survenant chez un petit enfant. Le pronostic vital est excellent dans les pays occidentaux, mais il paraît encore nécessaire d’améliorer le dépistage afin que la prise en charge de ces enfants soit la plus précoce possible. Les deux signes qui doivent alerter les professionnels de santé et faire réaliser un FO dans les meilleurs délais sont : la leucocorie et le strabisme.
Les enfants atteints de rétinoblastome nécessitent une prise en charge multidisciplinaire en milieu hautement spécialisé. Une fois les traitements terminés, un suivi ophtalmologique et général prolongé est nécessaire.
La consultation génétique est l’une des étapes indispensables, initialement pendant l’enfance puis à l’âge adulte, afin d’améliorer le taux de conservation oculaire, en préservant la fonction visuelle tout en limitant ou diminuant les effets secondaires et la toxicité potentielle des traitements.
Un livret d’information sur le rétinoblastome a été réalisé par l’association RETINOSTOP et est disponible sur le site Internet www.retinostop.org.
[1] Lacour B, Guyot-Goubin A, Guissou S, et al. Incidence of childhood cancer in France : National Children Cancer Registries, 2000–2004. Eur J Cancer Prev 2010 ; 19 : 173-81.
[2] Lumbroso-Le Rouic L, Savignoni A, Levy-Gabriel C, et al. Treatment of retinoblastoma : The Institut Curie experience on a series of 730 patients (1995 to 2009). J Fr Ophtalmol 2015 ; 38 : 535-41.
[3] Rothschild PR, Levy D, Savignoni A, et al. Familial retinoblastoma : fundus screening schedule impact and guideline proposal. A retrospective study. Eye (Lond) 2011 ; 25 : 1555-61.
[4] Vahedi A, Lumbroso-Le Rouic L, Levy Gabriel C, et al. Differential diagnosis of retinoblastoma: a retrospective study of 486 cases. J Fr Ophtalmol 2008 ; 31 : 165-72.
[5] Murphree AL. Intraocular retinoblastoma : the case for a new group classification. In : Ophthalmology clinics of north america. Vol. 18. Elsevier Saunders ; 2005.
[6] Sastre X, Chantada GL, Doz F, et al. Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med 2009 ; 133 : 1199-202.
[7] Aerts I, Sastre-Garau X, Savignoni A, et al. Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 2013 ; 31 : 1458-63.
[8] Lumbroso-Le Rouic L, Aerts I, Levy-Gabriel C, et al. Conservative treatments of intraocular retinoblastoma. Ophthalmology 2008 ; 115 : 1405-10, 1410.e1-2.
[9] Gobin YP, Dunkel IJ, Marr BP, et al. Intra-arterial chemotherapy for the management of retinoblastoma : four-year experience. Arch Ophthalmol 2011 ; 129 : 732-7.
[10] Munier FL, Beck-Popovic M, Balmer A, et al. Occurrence of sectoral choroidal occlusive vasculopathy and retinal arteriolar embolization after superselective ophthalmic artery chemotherapy for advanced intraocular retinoblastoma. Retina 2011 ; 31 : 566-73.
[11] Tsimpida M, Thompson DA, Liasis A, et al. Visual outcomes following intraophthalmic artery melphalan for patients with refractory retinoblastoma and age appropriate vision. Br J Ophthalmol 2013 ; 97 : 1464-70.
[12] Francis JH, Abramson DH, Gaillard MC, et al. The classification of vitreous seeds in retinoblastoma and response to intravitreal melphalan. Ophthalmology 2015 ; 122 : 1173-9.
[13] Desjardins L, Chefchaouni MC, Lumbroso L, et al. Functional results of retinoblastoma treatment with local treatment used in isolation or associated with chemotherapy. J Fr Ophtalmol 2005 ; 28 : 725-31.
[14] Aerts I, Pacquement H, Doz F, et al. Outcome of second malignancies after retinoblastoma : a retrospective analysis of 25 patients treated at the Institut Curie. Eur J Cancer 2004 ; 40 : 1522-9.