Opacités congénitales de cornée et dysgénésies du segment antérieur
Coordonné par E. Bui Quoc
E. Bui Quoc, M. Beylerian, D. Denis
Les interrogations sont multiples lorsqu’on aborde la question des opacités congénitales de la cornée et les dysgénésies du segment antérieur. En préambule, il faut souligner qu’elles demeurent des pathologies rares; ainsi, la fréquence des opacités congénitales de la cornée serait estimée à 3 naissances sur 100 000 [1].
Les deux sujets des opacités congénitales de cornée et des dysgénésies du segment antérieur peuvent concerner des pathologies communes, mais il existe des dysgénésies du segment antérieur dans lesquelles on ne retrouve pas d’opacité de cornée, comme il existe des opacités du segment antérieur qui ne sont pas des dysgénésies du segment antérieur (anomalies malformatives, constitutionnelles, innées) mais des pathologies acquises.
Dans ce caractère acquis, on peut retrouver une pathologie « constitutionnelle » . Ainsi, par exemple, le glaucome congénital entraîne dans sa forme précoce une opacité cornéenne uni- ou bilatérale, alors que dans sa forme tardive, il se caractérise par une opacification de la cornée plus progressive et différée.
Cette complexité de pathologies intriquées est en partie une des explications d’une nosologie complexe, ou plutôt variable, avec des classifications multiples des dysgénésies du segment antérieur, incluant des « anomalies » , des « syndromes » à noms propres multiples, décrivant des phénotypes variables. Les données anciennes, suite aux travaux d’excision graduée des cellules des crêtes neurales chez l’embryon de poulet par Johnston dans les années 1970, décrivant des anomalies de formation, migration, prolifération, différenciation, et intégrant les dysgénésies du segment antérieur dans le groupe « fourre-tout » des « neurocristopathies » , sont à réévaluer complètement à la lumière des données génétiques. Les « nouvelles » descriptions des dysgénésies du segment antérieur se veulent pragmatiques, distinguant des « opacités congénitales/néo-natales de la cornée » primaires et secondaires (Tableau 11-1) [2]. Dans les opacités primaires (le caractère étant en partie lié à l’atteinte endothéliale), on retrouve des causes innées (dystrophies endothéliales, dermoïdes du limbe, cornea plana) mais aussi le glaucome congénital par mutation de CYP1B1. Dans les opacités secondaires (une autre malformation se substituant ou se surajoutant à l’atteinte endothéliale), on retrouve des causes innées (dysgénésies du segment antérieur cornéo-irido-cristaliniennes ou iridotrabéculaires) ou acquises (traumatiques, métaboliques, infectieuses, etc.).
Notons d’ores et déjà qu’il existe un paradoxe nosologique du fait de l’antagonisme apparent entre les termes « congénital » et « acquis » . On « naît » avec une pathologie congénitale mais en réalité, dans des pathologies comme la cataracte congénitale, le glaucome congénital, ou même le « strabisme congénital » (ancienne appellation du strabisme précoce), il peut ne pas y avoir d’anomalie apparente à la naissance, mais la pathologie se développera car l’enfant est né avec le « programme » pathologique qui va conduire à la maladie (la génétique développementale est sous-jacente bien entendu). Dans les opacités « congénitales » acquises de la cornée, celles-ci peuvent également être présentes à la naissance ou survenir plus tard.
Outre le fait que dans cette classification, on inclut des pathologies dans lesquelles il n’existe pas d’opacité (Axenfeld par exemple), la génétique qui bouleverse nos points de vue phénotypiques de clinicien retrouve des gènes communs à des causes primaires et secondaires, mais aussi à des dysgénésies cornéo-irido-cristalliniennes et à des dysgénésies iridotrabéculaires. Ce qui signifie qu’il faut être pragmatique :
les classifications anciennes et nouvelles doivent être connues;
devant une pathologie, la description phénotypique doit être la plus précise possible :
dans un souci génotypique de recherche et de conseil génétique,
dans un souci thérapeutique : que faut-il faire? Que peut-on faire?
Ce chapitre fait le point sur : les questions de classification/nosologie; les génotypes et les corrélations génotype-phénotype; les questions pratiques de traitement médical et chirurgical; la difficile question de la greffe de cornée chez l’enfant; la prise en charge plus simple des dermoïdes du limbe.
Tableau 11-1 Opacités congénitales de cornée primaires et secondaires d’après Nischal [2].

CHED : congenital hereditary endothelial dystrophy ou dystrophie endothéliale congénitale héréditaire ; IC3D : classification de l’International Committee for Classification of Corneal Dystrophies ; KILD : kerato-irido-lenticular dysgenesis ou dysgénésie cornéo-irido- cristallinienne ; MIDAS : microphtalmia dermal aplasia sclerocornea ; PPCD : posterior polymorphous congenital dystrophy ou dystrophie postérieure polymorphe congénitale.
* Divers mécanismes conduisent à une KILD : adhésions iridocornéennes ; le cristallin ne se sépare pas de la cornée ; le cristallin se sépare de la cornée mais ne se forme pas correctement ; le cristallin ne se forme pas ; anomalies dues à une persistance de la vascularisation foetale/persistance du vitré primitif.
[1] Ciralsky J, Colby K Congenital comeal opacities : a review with a focus on genetics Semin Ophthalmol 2007 ; 22 : 241-246
[2] Nischal KK Genetics of congenital corneal opacification – impact on diagnosis and treatment Cornea 2015 ; 34 (Suppl 10) : S24-S34
P. Calvas, N. Chassaing
Le réseau transcriptionnel qui contrôle le développement oculaire n’est que partiellement déchiffré. De nombreuses molécules clés ont cependant été identifiées, en particulier près de 80 facteurs de transcription sont exprimés dans l’œil en développement. Parmi ceux-ci, une trentaine de gènes est jugée essentielle pour mettre en route ce programme de développement. Deux voies de signalisation semblent aujourd’hui indispensables aux étapes initiales d’induction de la différenciation oculaire. Les deux gènes majeurs qui y participent, PAX6 et SIX3, se disputent le titre de gène maître du développement de l’œil [1]. De nombreux autres gènes sont impliqués dans le développement des diverses structures oculaires. Les anomalies du segment antérieur sont dues à des mutations de gènes intervenant, en général, au cours de la différenciation terminale de l’œil. Ces défauts congénitaux mais évolutifs comprennent une grande variété d’anomalies élémentaires de la cornée, de l’iris et du cristallin, isolées ou associées entre elles. Les associations ont permis de définir certaines entités diagnostiques du fait de leur fréquence et de leur relative reproductibilité entre les patients, telles l’aniridie, l’anomalie d’Axenfeld-Rieger, l’anomalie de Peters…
Leur association à des manifestations viscérales extra-oculaires a de la même manière conduit à la description de syndromes tels le syndrome WAGR (Wilms tumor, Aniridia, Genital anomalies, mental Retardation), le syndrome d’Axenfeld-Rieger, le syndrome de Peters-plus (Krause-Kivlin) et d’autres entités plus rares ou dans lesquelles le phénotype oculaire n’est pas au premier plan (syndrome d’Alagille, syndrome SHORT pour Short stature, inguinal Hernia, Ocular depression, Rieger anomaly, delay in eruption of Teeth), etc.
De nombreux rapports permettent aujourd’hui de corréler certaines des anomalies cliniques avec le défaut de gènes du développement. Ces anomalies sont aussi variées que les anomalies oculaires et surtout les spectres cliniques se chevauchent. II s’ajoute donc une hétérogénéité génétique à l’hétérogénéité phénotypique. Ni la description anatomoclinique, ni la désignation d’un gène ne sont aujourd’hui suffisantes pour corréler le phénotype constaté avec l’anomalie d’un gène unique et aboutir à un conseil génétique approprié. Les différentes tentatives de classification des anomalies de développement du segment antérieur ne peuvent se contenter actuellement d’une définition clinique ou moléculaire exclusive et, de ce fait, leur complexité est croissante [2].
La détermination du gène responsable de la ou des mutations causales est cependant utile pour affirmer le diagnostic. Elle permet de mieux cerner le spectre phénotypique et le pronostic du cas index et des membres de sa famille. La définition d’un mode de transmission est, elle aussi, essentielle au conseil génétique. Les mutations des gènes responsables de dysgénésies oculaires sont très majoritairement dominantes. Cependant les histoires familiales ne traduisent pas toujours ce fait. D’une part, les nouvelles mutations sont nombreuses et le cas index est alors le premier et seul cas d’une famille, le risque de récurrence chez ses propres enfants est élevé. D’autre part, les mosaïques germinales ne sont pas exceptionnelles et il existe un risque pour des parents, non-porteurs de l’anomalie génétique présente chez leur enfant, d’assister à une récidive à l’occasion d’une nouvelle naissance.
Le lien entre les principaux gènes impliqués et les anomalies cliniques provoquées est présenté dans le Tableau 11-2. Quant au Tableau 11-3, il est à utiliser comme un guide synthétique des explorations génétiques à prévoir devant une association malformative. Les principaux gènes sont passés succinctement en revue ci-après.
La connaissance du lien entre les mutations du gène PAX6 et le développement d’anomalies oculaires date de 1992 [3]. Les phénotypes associés aux anomalies de ce gène ont été particulièrement étudiés et dépassent le cadre classique des aniridies [4, 5]. De nombreuses mutations de PAX6 aboutissent à une haplo-insuffisance (perte de fonction d’un allèle), ce qui explique l’absence générale de corrélation entre les génotypes et les phénotypes observés au sein du spectre des aniridies, aussi bien au sein d’une même famille qu’entre les familles partageant la même mutation. Les rares mutations faux-sens sont associées aux phénotypes variants exempts d’aniridie, comme les anomalies de Peters, d’Axenfeld-Rieger ou de la papille [6, 7], ou plus atténués [8]. Un nombre croissant de cas d’aniridie sans mutation retrouvée dans les séquences codantes du gène sont liés à des anomalies de la région régulatrice du gène située à son extrémité 3’ terminale; une analyse du gène doit comporter la recherche systématique d’une délétion de cette région avant d’éliminer sa responsabilité dans un phénotype compatible [9]. Dans notre expérience, ces délétions représentent environ 10 % des aniridies typiques sans anomalie retrouvée dans le gène PAX6, soit moins de 1 % des cas d’aniridie. Une mutation ponctuelle, unique, modifiant la séquence d’un élément majeur de régulation a été décrite démontrant que des analyses encore plus exhaustives du gène deviendront nécessaires avec leur facilitation par les techniques de séquençage à haut débit [10].
L’existence d’atteintes extra-oculaires se précise. Les délétions étendues du gène dans les syndromes WAGR [11] ou les rares formes d’hétérozygotie composite [12] ne sont plus les seules suspectes de provoquer des anomalies du développement cérébral et intellectuel [13]. Le syndrome de Gillespie qui associe à l’aniridie une ataxie cérebelleuse et un retard mental est un exemple qui sera traité avec les formes récessives autosomiques. Le suivi du développement neurosensoriel des enfants atteints d’aniridie est donc d’actualité et, notre équipe met en place une étude observationnelle dédiée : la cohorte nationale RaDiCo-AC-OEIL.
Les atteintes de la régulation glycémique font aussi l’objet d’un nombre croissant de publications et traduisent l’intérêt d’une surveillance glycémique chez les patients [14, 15].
Enfin, la mise en évidence récente d’anophtalmie ou microphtalmie sévère dans une forme hétérozygote de mutation de PAX6 traduit bien le continuum moléculaire existant entre les défauts d’induction de l’œil aboutissant à un arrêt complet de son développement et l’ensemble du contrôle de ce développement jusqu’à la différenciation terminale. Il se traduit par le même continuum clinique entre l’absence de globe oculaire et des défauts limités au segment antérieur [8, 16].
Tableau 11-2 Principaux gènes responsables de dysgénésies du segment antérieur de l’oeil et résumé des phénotypes associés aux mutations.

DA : dominant autosomique ; LX : lié à l’X ; RA : récessif autosomique ; SA : segment antérieur de l’oeil.
* Online Mendelian Inheritance in Man (OMIN®) : http://www.omim.org.
** Dysgénésie du SA : combinaison variable d’une opacité cornéenne, d’un embryotoxon postérieur, d’une hypoplasie irienne, d’une correctopie ou polycorie, de brides iridocornéennes ou entre le cristallin et la cornée. Axenfeld-Rieger : association d’une hypoplasie irienne, d’un embryotoxon postérieur, d’une correctopie ou polycorie et/ou de brides iridocornéennes. Peters : association d’une opacité cornéenne, d’une déhiscence de la membrane de Descemet et d’adhérences iridocornéeenne.
Tableau 11-3 Algorithme décisionnel schématique d’exploration génétique devant une anomalie congénitale du segment antérieur de l’oeil.
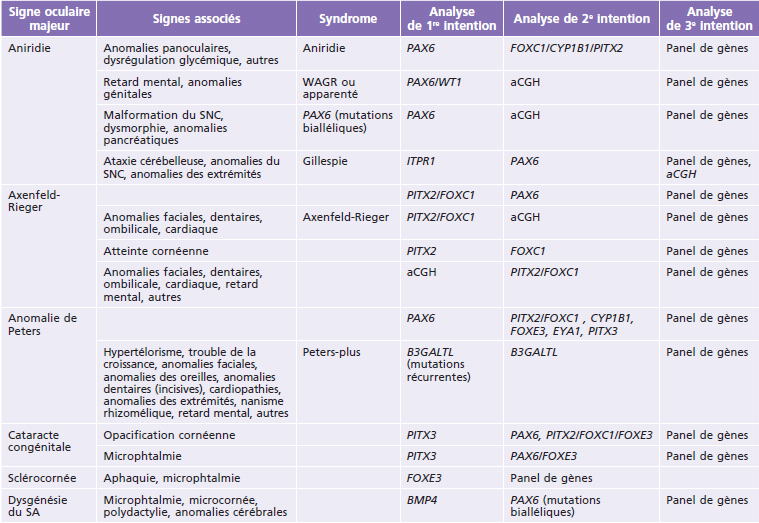
aCGH : hybridation génomique comparative sur puce à ADN ; SA : segment antérieur de l’oeil ; SNC : système nerveux central.
Ces deux gènes sont associés à l’anomalie d’Axenfeld-Rieger sans qu’une corrélation claire entre les génotypes de chacun d’eux et les phénotypes observés ne se dégage [17, 18]. Les mêmes gènes sont impliqués dans les syndromes d’Axenfeld-Rieger qui associent les composantes extra-oculaires faciales, dentaires, l’excès de peau péri-ombilical, les anomalies génitales et le retard de croissance. La variabilité de l’expression phénotypique inclut l’anomalie de Peters, les hypoplasies iriennes isolées qui sévères peuvent mimer l’aniridie, les anomalies de l’angle iridocornéen pourvoyeuses de glaucomes développementaux. Certaines anomalies sont rencontrées uniquement dans les mutations de PITX2 (dermoïdes cornéens) [19] ou de FOXC1 (aniridie et glaucome congénital). D’une manière générale, les anomalies de PITX2 sont plus souvent associées aux anomalies faciales, dentaires et de l’ombilic et celles de FOXC1 à une expression oculaire isolée ou à des malformations cardiaques. Cependant, l’existence exceptionnelle de formes très sévères de dysgénésies du segment antérieur fait que les deux analyses sont systématiquement associées au cours du diagnostic des anomalies évoquant l’implication de l’un ou l’autre de ces gènes [20]. Outre les anomalies intragéniques, de grands réarrangements génomiques régionaux (copy number variation ou CNV) peuvent exister : délétions de la région chromosomique 4q25 (PITX2) et délétions ou duplications de la région 6q25 (FOXC1) dont les phénotypes complexes dépendent de l’implication de régions régulatrices ainsi que de la nature et du nombre des gènes impliqués dans le CNV [21]. L’importante variabilité phénotypique a été soulignée dans de nombreux articles et est également illustrée par le recouvrement du syndrome d’Axenfeld-Rieger avec le syndrome SHORT [22].
Les mutations de ce gène sont une cause peu fréquente de cataractes congénitales polaires postérieures. L’association de ces dernières avec une dysgénésie du segment antérieur de l’œil doit amener à considérer également la responsabilité de ce gène [23]. Des formes plus sévères d’atteintes oculaires, incluant des microphtalmies et un retard de développement, sont décrites associées à des mutations homozygotes. La coexistence de formes sévères, associées à des mutations homozygotes ou à une atteinte de deux gènes intervenant dans la même voie de transcription (c’est-à-dire PITX2 et FOXC1) [20], reflète bien la complexité des réseaux transcriptionnels impliqués dans le développement. La variabilité phénotypique qui découle de l’atteinte d’un gène traduit le fait que celui-ci ne représente qu’un élément, souvent essentiel, mais ponctuel, de ce réseau. Une revue récente des phénotypes associés aux mutations de PITX3 illustre ce spectre phénotypique et les mécanismes impliqués [24]. On peut d’ailleurs noter un large territoire d’expression des gènes de la famille PITX. Par exemple, des analyses génomiques ont établi le rôle de PITX3 dans la maladie de Parkinson idiopathique [25], ceci venant illustrer le rôle du gène dans le développement cérébral et les conséquences de son dysfonctionnement qui peuvent être sévères.
La même complexité phénotypique est liée à la transmission du gène FOXE3 dont les mutations hétérozygotes composites sont responsables de microphtalmie-anophtalmie avec aphaquie et sclérocornée, et les mutations hétérozygotes sont responsables de dysgénésies du segment antérieur [26, 27]. Il est intéressant de noter que des mutations hétérozygotes particulières, supprimant le codon STOP du gène et conduisant à un allongement de la protéine, ou celles altérant la fonction de la partie terminale de la protéine sont responsables de phénotypes variants, proches de l’anomalie de Peters [28], ce que confirment nos observations personnelles.
Les mutations du gène BMP4 sont typiquement associées à des anomalies cérébrales, oculaires, craniofaciales, digitales et à un déficit de croissance [29]. Des formes moins sévères existent et le phénotype oculaire peut être limité à une dysgénésie du segment antérieur de l’œil. D’autres gènes de la famille BMP sont impliqués dans le développement oculaire, tel BMP7 qui participe au développement précoce et à celui des procès ciliaires et dont l’implication, probablement exceptionnelle, a été montrée dans des microphtalmies-anophtalmies syndromiques [30].
Des anomalies du segment antérieur sont présentes dans de nombreux syndromes transmis selon un mode dominant autosomique; ils sont très nombreux si l’on inclut les anomalies de l’angle irido-cornéen. Nous citerons, sans toutefois être exhaustif, le syndrome d’Alagille [31] et la maladie cérébrale microvasculaire liée aux mutations de COL4A1 dans laquelle l’implication du segment antérieur est fréquente et variée [32, 33].
Nous avons vu avec l’exemple de FOXE3 que des gènes dont les mutations sont majoritairement dominantes peuvent aussi héberger des mutations récessives. Ceux qui suivent n’ont en l’état actuel des connaissances que peu ou pas d’effet sur les porteurs hétérozygotes et seules les atteintes bialléliques donnent lieu à des dysgénésies du segment antérieur. Ces anomalies sont comparables à celles déjà évoquées plus haut.
Ses mutations homozygotes ou hétérozygotes composites sont associées au glaucome congénital primitif [34]. Son implication recouvre aussi les anomalies et syndromes d’Axenfeld-Rieger [35].
B3GALTL est à ce jour le seul gène impliqué dans le syndrome de Peters-plus. Il s’agit d’un syndrome rare qui associe un défaut du segment antérieur (typiquement une anomalie de Peters), un déficit de croissance, une brachydactylie, un raccourcissement rhizomélique, une dysmorphie craniofaciale, une fente labiopalatine, des anomalies cardiaques et génito-urinaires. Le syndrome a été bien délimité par la publication de Maillette de Buy Wenniger-Prick et al. [36]. Seules les formes complètes sont associées aux mutations de B3GALTL. Certaines mutations sont récurrentes, ce qui en facilite la détection, et la plupart des mutations rencontrées sont des mutations d’épissage [37]. Les mutations du gène sont en revanche absentes dans les formes atypiques et leur recherche n’est pas indiquée. Al-Gazali et al. [38] ont décrit l’absence de mutation du gène dans une forme complète et sévère questionnant l’existence d’un nouveau syndrome ou d’une hétérogénéité génétique.
Comme pour les formes à transmission dominante, plusieurs syndromes peuvent de façon plus occasionnelle s’accompagner de dysgénésies du segment antérieur, mais leur diagnostic est plus dépendant des signes majeurs ou plus constants. La délimitation du rôle des gènes en cause, impliqués dans le développement oculaire, demeure importante mais relève du domaine de l’investigation plutôt que de la clinique.
Le syndrome de Gillespie est une affection rare qui associe à une aniridie, une ataxie cérébelleuse, des anomalies des extrémités et un retard de développement. Plusieurs modes de transmission ont été évoqués : récessif autosomique, dominant autosomique et lié à l’X [39]. La recherche des gènes responsables, peu fructueuse, traduisait une probable hétérogénéité génétique et une définition clinique complexe ou hétérogène. On a pu tour à tour incriminer le gène PAX6 puis le dédouaner [40, 41]. Très récemment, en utilisant des analyses de l’exome entier, Gerber et al. [42] et McEntagart et al. [43] ont mis en évidence des mutations dominantes et récessives du gène IPTR1 comme étant à l’origine de l’ensemble des cas index porteurs d’un syndrome de Gillespie. Tous les patients présentaient un phénotype clinique et une imagerie cérébrale homogènes, ce qui conforte ITPR1 comme le gène majoritaire du syndrome. Le débat reste cependant ouvert quant à l’existence d’une hétérogénéité génétique à l’origine des aniridies et du syndrome de Gillespie [39].
La plupart des gènes intervenant dans la différenciation terminale du segment antérieur de l’œil sont impliqués dans la genèse des dysgénésies oculaires. Il s’agit en majorité de facteurs de transcription qui possèdent aussi une expression extra-oculaire. Celle-ci explique l’existence de formes syndromiques et une part de la variabilité phénotypique intra- et extrafamiliale. Des gènes majeurs du développement précoce de l’œil peuvent aussi être impliqués dans des défauts de la différenciation terminale, c’est très régulièrement le cas de PAX6 ou exceptionnellement celui de SOX2. En complément à la mise en évidence de mutation de ces gènes, les techniques actuelles d’hybridation génomique comparative ont permis d’étendre la recherche des causes génétiques aux CNV régionales et de mettre en évidence des anomalies complexes impliquant les gènes connus ou leurs séquences régulatrices. Elles ont permis de découvrir de nouvelles régions candidates à la genèse des phénotypes oculaires explorés. L’évolution des moyens de détection des mutations des gènes avec le passage du séquençage à moyen débit et rapidement à haut débit en routine diagnostique enrichira très certainement ces connaissances.
[1] Sinn R, Wittbrodt J An eye on eye development Mech Dev 2013 ; 130 : 347-358
[2] Alward WL Axenfeld-Rieger syndrome in the age of molecular genetics Am J Ophthalmol 2000 ; 130 : 107-115
[3] Jordan T, et al. The human PAX6 gene is mutated in two patients with aniridia Nat Genet 1992 ; 1 : 328-332
[4] Prosser J, Van Heyningen V PAX6 mutations reviewed Hum Mutat 1998 ; 11 : 93-108
[5] Vincent MC, et al. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects Eur J Hum Genet 2003 ; 11 : 163-169
[6] Hanson I, et al. Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations Hum Mol Genet 1999 ; 8 : 165-172
[7] Azuma N, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations Am J Hum Genet 2003 ; 72 : 1565-1570
[8] Beby F, Dieterich K, Calvas P A [c.566-2A>G] heterozygous mutation in the PAX6 gene causes aniridia with mild visual impairment Eye (Lond) 2011 ; 25 : 657-658
[9] Lauderdale JD, et al. 3’ deletions cause aniridia by preventing PAX6 gene expression Proc Natl Acad Sci U S A 2000 ; 97 : 13755-13759
[10] Bhatia S, et al. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia Am J Hum Genet 2013 ; 93 : 1126-1134
[11] Yamamoto T, et al. Narrowing of the responsible region for severe developmental delay and autistic behaviors in WAGR syndrome down to 1.6 Mb including PAX6, WT1, and PRRG4 Am J Med Genet A 2014 ; 164A : 634-638
[12] Glaser T, et al. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects Nat Genet 1994 ; 7 : 463-471
[13] Yogarajah M, et al. PAX6, brain structure and function in human adults : advanced MRI in aniridia Ann Clin Transl Neurol 2016 ; 3 : 314-330
[14] Laakso M Not for the eyes only : PAX6 and glucose metabolism Diabetologia 2009 ; 52 : 381-384
[15] Gosmain Y, et al. Pax6 controls the expression of critical genes involved in pancreatic {alpha} cell differentiation and function J Biol Chem 2010 ; 285 : 33381-33393
[16] Chassaing N, et al. Targeted resequencing identifies PTCH1 as a major contributor to ocular developmental anomalies and extends the SOX2 regulatory network Genome Res 2016 ; 26 : 474-485
[17] Semina EV, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome Nat Genet 1996 ; 14 : 392-399
[18] Nishimura DY, et al. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25 Nat Genet 1998 ; 19 : 140-147
[19] Xia K, et al. Mutation in PITX2 is associated with ring dermoid of the cornea J Med Genet 2004 ; 41 : e129
[20] Kelberman D, et al. Digenic inheritance of mutations in FOXC1 and PITX2 : correlating transcription factor function and Axenfeld-Rieger disease severity Hum Mutat 2011 ; 32 : 1144-1152
[21] D’Haene B, et al. Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations Invest Ophthalmol Vis Sci 2011 ; 52 : 324-333
[22] Karadeniz NN, et al. Is SHORT syndrome another phenotypic variation of PITX2? Am J Med Genet A 2004 ; 130A : 406-409
[23] Semina EV, et al. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD Nat Genet 1998 ; 19 : 167-170
[24] Medina-Martinez O Focus on molecules : PITX3 Exp Eye Res 2012 ; 99 : 106-107
[25] Smidt MP, Smits SM, Burbach JP Homeobox gene PITX3 and its role in the development of dopamine neurons of the substantia nigra Cell Tissue Res 2004 ; 318 : 35-43
[26] Valleix S, et al. Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans Am J Hum Genet 2006 ; 79 : 358-364
[27] Chassaing N, et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia Clin Genet 2013 ; 86 : 326-334
[28] Doucette L, et al. A novel, non-stop mutation in FOXE3 causes an autosomal dominant form of variable anterior segment dysgenesis including Peters anomaly Eur J Hum Genet 2011 ; 19 : 293-299
[29] Reis LM, et al. BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome Hum Genet 2011 ; 130 : 495-504
[30] Wyatt AW, et al. Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies Hum Mutat 2010 ; 31 : 781-787
[31] Hingorani M, et al. Ocular abnormalities in Alagille syndrome Ophthalmology 1999 ; 106 : 330-337
[32] van der Knaap MS, et al. Neonatal porencephaly and adult stroke related to mutations in collagen IV A1 Ann Neurol 2006 ; 59 : 504-511
[33] Sibon I, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke Ann Neurol 2007 ; 62 : 177-184
[34] Stoilov I, Akarsu AN, Sarfarazi M Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21 Hum Mol Genet 1997 ; 6 : 641-647
[35] Cella W, et al. Structural assessment of PITX2, FOXC1, CYP1B1, and GJA1 genes in patients with Axenfeld-Rieger syndrome with developmental glaucoma Invest Ophthalmol Vis Sci 2006 ; 47 : 1803-1809
[36] Maillette de Buy Wenniger-Prick LJ, Hennekam RC The Peters’ plus syndrome : a review Ann Genet 2002 ; 45 : 97-103
[37] Reis LM, et al. Mutation analysis of B3GALTL in Peters Plus syndrome Am J Med Genet A 2008 ; 146A (20) : 2603-2610
[38] Al-Gazali L, et al. Anterior segment anomalies of the eye, growth retardation associated with hypoplastic pituitary gland and endocrine abnormalities : Jung syndrome or a new syndrome? Am J Med Genet A 2009 ; 149A (2) : 251-256
[39] Ansari M, et al. Genetic analysis of ‘PAX6-Negative’ individuals with aniridia or Gillespie syndrome PLoS One 2016 ; 11 : e0153757
[40] Ticho BH, et al. Ocular findings in Gillespie-like syndrome : association with a new PAX6 mutation Ophthalmic Genet 2006 ; 27 : 145-149
[41] Wittig EO, et al. Partial aniridia, cerebellar ataxia, and mental deficiency (Gillespie syndrome) in two brothers Am J Med Genet 1988 ; 30 : 703-708
[42] Gerber S, et al. Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome Am J Hum Genet 2016 ; 98 : 971-980
[43] McEntagart M, et al. A restricted repertoire of de novo mutations in ITPR1 cause Gillespie syndrome with evidence for dominant-negative effect Am J Hum Genet 2016 ; 98 : 981-992
E. Bui Quoc, M. Beylerian, D. Denis
En ophtalmologie pédiatrique, le terme « congénital » est rarement adapté à la pathologie décrite, que ce soit pour le glaucome congénital, la cataracte congénitale ou les opacités congénitales de la cornée, etc. L’adjectif n’est pas approprié, car il ramène à la notion de « naître avec » (du latin congenitus). Or, la pathologie ne s’exprime pas forcément en post-natal immédiat mais peut se manifester plus tardivement. Il faut donc parfois comprendre le terme « congénital » comme ramenant à une notion de pathologie innée, à la différence d’une pathologie acquise. Dans les « opacités congénitales de la cornée » , il existe à la fois des causes innées malformatives, déterminées par une mutation de gènes du développement de l’œil (connus ou pas), mais aussi des causes acquises exogènes ou endogènes (infectieuses, métaboliques, traumatiques).
L’œil est une structure dont les éléments se mettent en place très tôt au cours de l’embryogenèse, avec un déterminisme conduit par des gènes du développement, intervenant selon une chronologie précise, dans les premières semaines de vie embryonnaire. Rappelons très brièvement une notion classique : le système nerveux central se développe à partir de la plaque neurale à la 3e semaine de vie embryonnaire, avec une invagination pour former la gouttière neurale, laquelle présente deux « crêtes » . Les cellules de la crête neurale constituent notamment les structures du segment antérieur de l’œil, d’où l’ancien terme de « neurocristopathie » pour décrire les pathologies de structures dérivées de la crête neurale (voir chapitre 28.1).
Un peu d’histoire : des travaux princeps datant des années 1970, sur l’excision graduée de crêtes neurales chez l’embryon de poulet, ont montré selon le moment de l’excision que les anomalies oculaires induites étaient variables [1]. Selon l’atteinte, les différentes anomalies malformatives du segment antérieur décrites résultent d’anomalies de formation, de migration, de prolifération ou de différenciation des cellules dérivées des crêtes neurales :
l’anomalie de formation entraîne la cyclopie (formation d’une seule vésicule optique ne donnant qu’un seul œil); cette anomalie n’est pas viable;
les anomalies de migration des cellules de la crête neurale entraînent :
le glaucome congénital primitif,
les dysgénésies du segment antérieur : embryotoxon postérieur, anomalie d’Axenfeld, anomalie de Peters, anomalie de Rieger, sclérocornée, aniridie (concerne l’ensemble des tissus oculaires);
les anomalies de prolifération entraînent : atrophie essentielle de l’iris, syndrome de Chandler, syndrome de l’iris nævus;
la dystrophie postérieure polymorphe de la cornée est la conséquence d’une anomalie de différenciation des cellules de la crête neurale.
Plus tard, selon une approche plus anatomique et plus clinique (moins théorique), Hoskins, Shaffer et Hetherington ont proposé une autre classification des dysgénésies du segment antérieur, distinguant [2] :
le glaucome congénital primitif isolé constituant une trabéculodysgénésie isolée (anomalie de développement de l’angle);
les trabéculodysgénésies associées où il peut exister un glaucome par anomalie du développement de l’œil et du segment antérieur :
l’embryotoxon postérieur constituant une cornéodysgénésie, – l’aniridie et l’ectropion congénital de l’épithélium pigmenté constituant une trabéculo-irido-dysgénésie,
les anomalies d’Axenfeld, de Rieger ou de Peters constituant les trabéculo-cornéo-irido-dysgénésies.
Il est aisé de comprendre ou de formuler l’hypothèse que les malformations du segment antérieur de l’œil sont les conséquences d’anomalies du développement du segment antérieur et de la maturation de l’œil, avec des tableaux cliniques dépendant du moment où les anomalies (des gènes du développement de l’œil) s’expriment.
De fait, la génétique nous enseigne que ce sont des gènes du développement oculaire qui – du fait des mutations, dominantes autosomiques dans certains cas, récessives autosomiques dans d’autres cas – sont la cause des dysgénésies du segment antérieur et de certaines opacités congénitales de la cornée (voir GÉnÉTIQUE DES DYSGÉNÉSIES DU SEGMENT ANTERIEUR DE L‘ŒIL). La caractérisation génotypique montre que de nombreux gènes sont en cause : CYP1B1, PAX6, PITX2, FOXC1, etc. et que la question des corrélations phénotypes/génotypes est complexe, puisqu’un gène peut être responsable (selon les mutations causales) de plusieurs phénotypes et qu’un même phénotype peut être la conséquence de mutations de différents gènes. C’est pourquoi de nouvelles classifications des dysgénésies du segment antérieur et des anomalies de transparence de l’œil, primaires et secondaires, innées et acquises, ont été proposées (voir Tableau 11-1 et chapitre 9.1) [3]. Cette classification est descriptive et présente les dysgénésies du segment antérieur et les opacités congénitales/néonatales de cornée, primaires ou secondaires.
Pour rappel, dans les opacités primaires qui sont liées à une atteinte endothéliale, on retrouve des causes innées : dystrophie endothéliale, dermoïde du limbe, cornea plana, sclérocornée périphérique et cytopathie CYP1B1.
Dans les opacités secondaires, les causes sont :
innées :
dysgénésies cornéo-irido-cristaliniennes, dans lesquelles différents processus se produisent (adhésions iridocornéennes, absence de séparation entre cristallin et cornée, séparation entre cristallin et cornée mais malformation du cristallin, absence de formation du cristallin = aphaquie congénitale, malformations secondaires à des anomalies de la vascularisation fœtale); on retrouve dans ce groupe des phénotypes variables : les anomalies de Peters, les sclérocornées, le staphylome du segment antérieur, l’aphaquie congénitale, etc.;
dysgénésies iridotrabéculaires : anomalies d’Axenfeld et d’Axenfeld-Rieger, aniridie, glaucome infantile ou congénital, isolé ou secondaire à la dysgénésie;
acquises : traumatiques, métaboliques, infectieuses, etc.
Quelle que soit la « case nosologique » dans laquelle une anomalie dysgénésique ou une opacité congénitale de la cornée est classée, une des difficultés est que chaque clinicien peut évoquer un nom qui revêt pour les autres une signification différente. Une homogénéisation des diagnostics et des phénotypes doit se poursuivre, avec une description uniquement clinique et des questions simples à se poser : où se situe l’opacité de cornée? Comment est l’angle? Y a-t-il un glaucome associé? Comment sont l’endothélium, l’iris, le cristallin et le reste de l’anatomie oculaire? Quel est le pronostic anatomique et fonctionnel?
Les anomalies primitives de l’endothélium (anomalie de différentiation et/ou de migration des cellules endothéliales) sont responsables de tableaux multiples [4, 5]. La meilleure connaissance des génotypes distingue (voir Tableau 11-1) :
les dystrophies endothéliales cornéennes héréditaires;
les dystrophies postérieures polymorphes congénitales.
Des phénotypes très rares ont également été décrits, comme la dystrophie postérieure amorphe congénitale (indiquée dans la classification de l’International Committee for Classification of Corneal Dystrophies ou IC3D dans les dystrophies stromales; voir chapitre 9) et la dystrophie cornéenne endothéliale liée à l’X
Dans les dystrophies endothéliales cornéennes héréditaires (corneal hereditary endothelium dystrophy [CHED]), les anomalies primitives de l’endothélium cornéen, mises en évidence en microscopie spéculaire, retrouvent une diminution de la densité et un pléomorphisme de ces cellules. Le glaucome associé est la conséquence d’une prolifération anormale des cellules endothéliales avec migration dans l’angle et sécrétion de collagène, avec obstruction de l’angle. L’œdème cornéen survient lorsque la densité cellulaire est inférieure à une densité critique (500 cellules environ). La distinction en CHED-1 et CHED-2 est remise en cause, avec exclusion de la CHED-1 faute de preuve de la transmission dominante, le CHED-2 devenant l’unique « CHED » .
Classiquement, dans le type 1, le début de la maladie survient tôt, vers 2 ans, avec évolutivité et opacification de la cornée entre 5 et 10 ans. La transmission est dominante autosomique avec mutation du gène PPCD1, situé dans la région péricentromérique du chromosome 20 (20p11.2q11.2), dans une zone que partage un gène responsable d’un type de dystrophie cornéenne postérieure polymorphe.
Dans le type 2, il existe une atteinte congénitale ou néonatale, avec un œdème cornéen stromal précoce, avec nystagmus et malvoyance précoces. La transmission est récessive autosomique par mutations homozygotes du gène SLC4A11. La pathologie est relativement stable.
Dans les dystrophies postérieures polymorphes congénitales (posterior polymorphous congenital dystrophy [PPCD]), les anomalies sont plus limitées que dans les CHED, avec présence d’agrégats de vésicules au niveau de la membrane de Descemet, limitant peu la vision et sans œdème cornéen. Différents gènes seraient impliqués avec un mode de transmission dominant autosomique : PPCD1 : gène VSX1; PPCD2 : gène COL8A2; PPCD3 : gène ZEB1.
Dans la dystrophie postérieure amorphe (posterior amorphous congenital dystrophy [PACD]), il existe une opacification lamellaire partielle ou complète de la cornée, une diminution de l’épaisseur de la cornée avec aplatissement de la courbure cornéenne. L’analyse génétique retrouve une délétion dans une zone codant pour des protéoglycanes [6].
La dystrophie cornéenne endothéliale liée à l’X est rare; il existe un œdème postérieur de la cornée; le traitement peut requérir une greffe de cornée (voir chapitre 9.1).
Voir chapitre 11.6.
La sclérocornée périphérique ou cornea plana résulte d’un arrêt du développement au cours du 4e mois d’âge fœtal, entraînant un aplatissement du rayon de courbure cornéen, inférieur à 43 dioptries. Il existe une forme dominante et une forme récessive qui partagent comme aspects cliniques : une réduction du rayon de courbure de la cornée et un limbe indistinct à un âge précoce. La forme autosomique dominante est la forme CNA1 qui se caractérise par une réduction du rayon de courbure de la cornée, une hypermétropie et une sclérocornée périphérique. Alors que la forme CNA2 (de transmission autosomique récessive par mutation du gène KERA situé en 12q22 et codant pour une protéine « keratocan » qui joue un rôle dans la transparence de la cornée) se caractérise par une réduction du rayon de courbure de la cornée, une microcornée, une sclérocornée périphérique et un œdème de cornée central avec opacités stromales.
Dans cette forme rare d’opacité congénitale de la cornée, celle-ci est due à la fois à l’œdème stromal du glaucome, mais aussi à l’absence d’endothélium et de Descemet. La greffe de cornée peut être requise mais le glaucome demeure de traitement difficile [3].
Les phénotypes sont divers selon les anomalies de formation et de séparation entre iris, cornée et cristallin. Ces dysgénésies cornéo-irido-cristalliniennes (kerato-irido-lenticular dysgenesis ou KILD) prennent des formes variées selon les adhésions de l’iris et de la cornée, la séparation complète ou incomplète du cristallin de la cornée, la malformation ou l’absence de formation du cristallin, ou du fait que ces anomalies sont secondaires à une persistance de la vascularisation fœtale.
Classiquement, dans ce qu’on appelle ou appelait l’anomalie de Peters, une classification propose trois groupes :
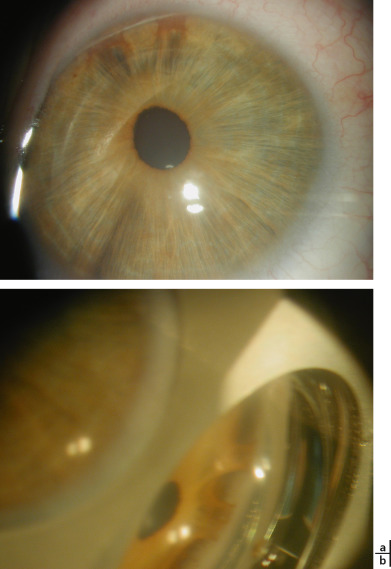
leucome cornéen central isolé dÛ à un amincissement cornéen postérieur. Le cristallin et l’iris ne présentent aucune anomalie (Fig. 11-1) = anomalie de Peters de type 1 selon certaines classifications;
leucome cornéen majeur avec adhérences iridocornéennes caractéristiques partant de la collerette irienne pour s’insérer à la face postérieure de la cornée (Fig. 11-2a, b) = anomalie de Peters de type 1 selon certaines classifications;
leucome cornéen avec adhérences iridocornéennes et adhérences cornéocristalliniennes avec un cristallin proche de la face postérieure de la cornée et/ou un cristallin subluxé et/ou cataracté (Fig. 11-2c) = anomalie de Peters de type 2 selon certaines classifications. L’ultrasound biomicroscopy (UBM) aide au diagnostic (Fig. 11-2d).
Les adhérences cornéocristalliniennes peuvent être décrites en plusieurs types :
le cristallin peut être adhérent au stroma cornéen avec absence de membrane de Descemet et de capsule lenticulaire;
le cristallin peut être localisé dans une position lointaine mais seulement apposé et non adhérent à la surface postérieure de la cornée;
le cristallin peut être en place mais avec une portion de la capsule antérieure et du cortex cristallinien en contact avec la face postérieure de la cornée;
le cristallin peut être en place mais cataracté en forme de cône avec un défect cornéen postérieur;
le cristallin peut être en place mais présenter une cataracte polaire antérieure ou une cataracte nucléaire.
L’UBM et/ou la tomographie par cohérence optique (optical coherence tomography [OCT]) du segment antérieur sont donc indispensables au diagnostic pour permettre un bilan exact des adhérences entre l’iris et la cornée et/ou entre la cornée et le cristallin.
L’anomalie de Peters peut s’associer à des anomalies systémiques. Par exemple, l’association syndromique dénommée « Peters-plus » [7] associe une atteinte cardiaque inconstante (foramens auriculaire ou ventriculaire perméables, sténose de la valve aortique, sténose de l’artère pulmonaire, bicuspidie de la valve pulmonaire) et une petite taille. Il existe une dysmorphie faciale, une fente labiale dans près de la moitié des cas, une fente palatine dans un tiers des cas, la présence de fistules pré-auriculaires. Des anomalies génito-urinaires ont été décrites plus rarement. Une atteinte cérébrale associée peut montrer une agénésie du corps calleux, une hydrocéphalie, un certain degré de microcéphalie, etc. La génétique a bien identifié le gène responsable de ce syndrome, puisque la plupart des sujets atteints sont homozygotes pour une mutation de l’intron 8 (c.660+ 1G>A) du gène B3GALTL [8].
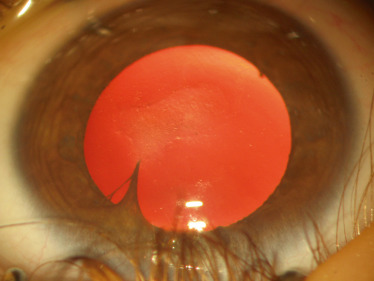
Fig. 11-1 Leucome central et synéchie iridocornéenne.
L’anomalie de Peters de type 1 se caractérise par une opacification plus ou moins vaste de la cornée (Fig. 11-1 et Fig. 11-2a), au niveau d’une discontinuité dans la membrane de Descemet et dans celle de l’endothélium [9]. Il existe, au niveau de l’opacité, des synéchies iridocornéennes constituées de ponts iriens partant de la pupille vers le lieu de la discontinuité cornéenne interne. Ces synéchies sont visibles en UBM et/ou après dilatation irienne (Fig. 11-2b). La présence d’un croissant cornéen clair périphérique permet le diagnostic différentiel avec un glaucome congénital. L’anomalie de Peters peut être uni- ou bilatérale. On peut également retrouver une anomalie de Peters de type 1 sur un œil et une autre dysgénésie du segment antérieur sur l’autre œil (par exemple : aniridie orientant, dans ce cas, vers une mutation de PAX6 ou anomalie d’Axenfeld-Rieger orientant vers une mutation de PITX2). De fait, le génotype dans l’anomalie de Peters de type 1 est variable : hérédité autosomale dominante ou récessive, mutations de gènes multiples : PAX6, CYP1B1, PITX2 (RIEG1), PITX3, FOXE3 ou FOXC1, etc. L’anomalie de Peters de type 1 peut s’associer à un glaucome qui sera dans ce cas précoce. Une implication de CYP1B1 a été démontrée dans ces cas. Des études histopathologiques ont montré la présence de synéchies périphériques antérieures. Des études ultrastructurales ont révélé des altérations trabéculaires.
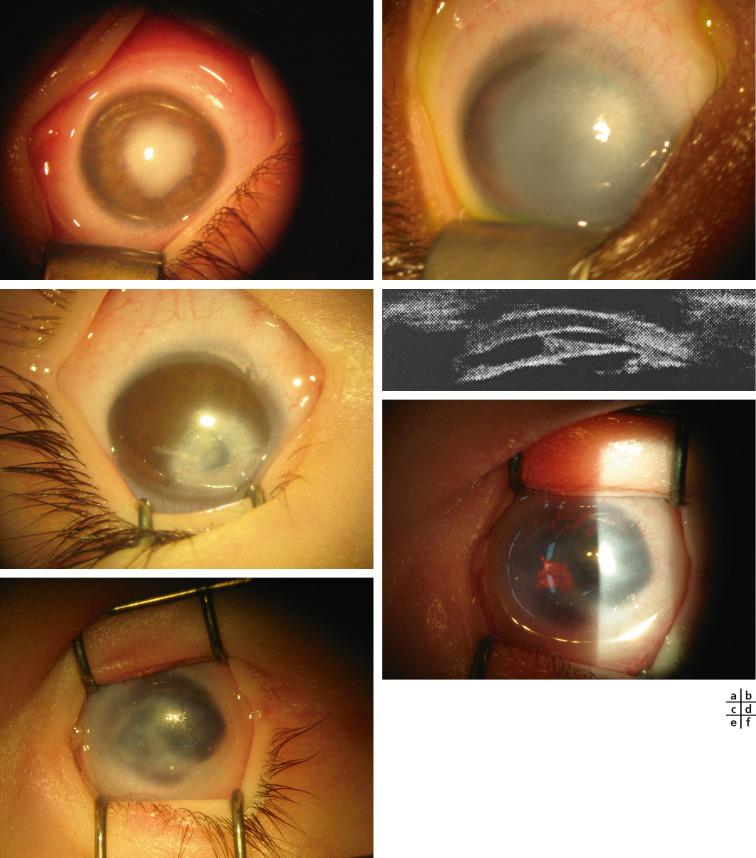
La sévérité de l’atteinte dans le cadre d’une anomalie de Peters peut varier, d’où la classification en trois stades évoquée plus haut. Lorsque l’atteinte se caractérise par des synéchies dans l’angle (entraînant volontiers un glaucome et une distension du globe), l’opacité centrale de la cornée s’associe à une anomalie cornéenne. L’anomalie cornéenne ne se réduit pas à une discontinuité de l’endothélium et de la membrane de Descemet, mais il peut exister dans les formes sévères un véritable amincissement central de la cornée avec risque de perforation (les structures iriennes basculent en avant derrière la cornée, la chambre antérieure est effacée et le cristallin vient s’apposer à la face postérieure de la cornée). Il s’agit de l’anomalie de Peters de type 2, de gravité variable : limitée (Fig. 11-2c) ou sévère (forme parfois nommée anomalie de von Hippel dans la littérature francophone) (Fig. 11-2). Le glaucome associé à cette malformation sévère de l’œil est donc d’une physiopathologie double : blocage pupillaire et malformation trabéculaire. Le pronostic fonctionnel est extrêmement compromis dans les formes sévères.
La sclérocornée entraîne, comme l’anomalie de Peters, une perte de transparence de la cornée qui devient complètement blanche, à l’instar de la sclère, avec un aspect vascularisé (Fig. 11-3). Il n’existe pas d’espace clair entre la sclère « normale » et la cornée opacifiée, ce qui la différencie de l’anomalie de Peters. La physiopathologie est différente et on ne retrouve pas de synéchies iridocornéennes en UBM. Il s’agit d’une non-organisation des fibres de collagène de la cornée qui ne prennent pas une disposition régulière et parallèle, permettant la transparence optique. Un très discret éclaircissement survient avec les années, mais sans permettre une vision chiffrable. Le diagnostic différentiel avec un glaucome congénital (lequel peut être associé) est aisé, car il n’y a ni buphtalmie ni œdème de cornée. La sclérocornée peut être partielle avec une zone claire centrale plus ou moins grande. Le gène KERA a été mis en cause dans la sclérocornée.
On distingue selon l’importance de l’opacité trois types cliniques :
type I (80 % ) : atteinte périphérique associée à une cornea plana (voir plus loin);
type II : atteinte périphérique ou centrale avec microphtalmie;
type III : atteinte périphérique et moyenne seulement.
Le staphylome congénital est une forme sévère de dysgénésie du segment antérieur (Fig. 11-4). C’est une anomalie unilatérale ou bilatérale rare qui représenterait environ 10 % des opacités cornéennes congénitales [10]. Les signes cliniques sont une cornée élargie, vascularisée, opaque, ectasique avec un amincissement et une ectasie des structures adjacentes du segment antérieur. Les anomalies du segment antérieur peuvent inclure un iris partiellement absent ou adhérent à la surface postérieure de la cornée. Le cristallin peut être adhérent à la surface de la cornée postérieure (similaire à l’anomalie de Peters), être subluxé, cataracté ou absent. Le degré de sévérité du staphylome peut être variable. Les staphylomes limbiques se produisent en raison de l’étirement et de l’amincissement du globe oculaire, secondaire à la pression intra-oculaire élevée, témoignant du glaucome congénital sévère. Plusieurs facteurs étiologiques ont été impliqués dans la pathogenèse du staphylome congénital antérieur : infection intra-utérine, anomalies chromosomiques avec d’autres atteintes organiques, association au syndrome de brides amniotiques [11]. Des mutations de CYP1B1 ont été identifiées dans des cas de staphylome congénital du segment antérieur [12].
Dans l’aphaquie congénitale, la cornée prend un aspect gris éclatant, avec diagnostic positif par l’échographie qui ne retrouve pas de cristallin.
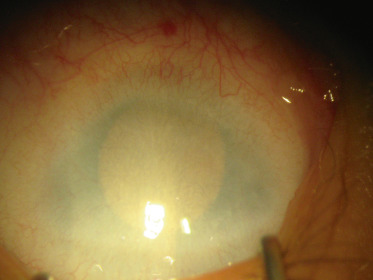
Fig. 11-3 Sclérocornée.
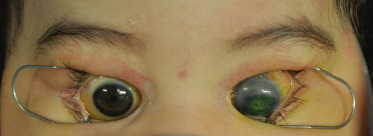
Fig. 11-4 Staphylome du segment antérieur gauche.
Les phénotypes secondaires à la persistance de la vascularisation fœtale/persistance du vitré primitif sont variables selon la sévérité de l’atteinte.
L’anomalie d’Axenfeld est caractérisée par des adhérences entre l’iris et la cornée au niveau de l’angle, entraînant une déformation parfois discrète de la pupille (Fig. 11-5a) dont la présence doit faire suspecter le diagnostic et faire réaliser une gonioscopie mettant en évidence de véritables ponts tissulaires périphériques entre l’iris et la cornée (Fig. 11-5b). Cette anomalie est un facteur de risque de glaucome précoce et requiert une surveillance régulière afin de dépister un glaucome débutant. Il existe un embryotoxon postérieur, c’est-à-dire un épaississement et un déplacement antérieur de la ligne de Schwalbe, sous forme d’une ligne blanc grisâtre en anneau parallèle au limbe, qui peut apparaître comme une fine ligne blanche isolée ou très épaissie et/ou déplacée en avant, suspendue par une fine membrane à la face postérieure de la cornée. L’embryotoxon postérieur isolé n’est pas aussi fréquent que le suggère la littérature ancienne. Il constituerait une anomalie d’Axenfeld a minima. Il impose dans tous les cas une surveillance régulière du tonus oculaire. Le mode de transmission de l’anomalie d’Axenfeld est autosomique dominant avec pénétrance complète et expressivité variable; les principaux gènes rencontrés sont PITX2 (4q25) et FOXC1 (6p25) dans plus de 50 % des cas, mais aussi PAX6 (11p13), gènes homéogènes qui codent pour des facteurs de transcription et orchestrent le développement des cellules de la crête neurale.
L’anomalie d’Axenfeld-Rieger ou anomalie de Rieger est une dysgénésie du segment antérieur dans laquelle les caractères de l’embryotoxon et de l’anomalie d’Axenfeld s’associent à une hypoplasie irienne, à une atrophie focale de l’iris (parfois improprement dénommée polycorie) et à un ectropion de l’uvée. Ces anomalies sont évolutives avec risque de glaucome. L’anomalie d’Axenfeld-Rieger est bilatérale, mais parfois asymétrique et le glaucome est associé dans la moitié des cas. Les autres signes oculaires associés sont : une microsphérophaquie, une microphtalmie, une microcornée, un staphylome, une sclérocornée et un strabisme. L’hérédité est autosomique dominante, avec des mutations des gènes FOXC1 et PITX2 [13].
Lorsque des anomalies systémiques sont présentes, on parle de syndrome de Rieger ou syndrome d’Axenfeld-Rieger, caractérisé par différentes malformations [14] :
dentaires : absence des dents ou microdontie intéressant surtout les incisives supérieures;
faciales : hypoplasie de la branche montante du maxillaire inférieur, aplatissement de la base du nez et hypertélorisme, lèvre inférieure proéminente;
ombilicales : défaut de régression du tissu péri-ombilical, confondu avec une hernie ombilicale;
urogénital : hypospadias;
osseuses : anomalies des hanches;
ORL : surdité;
cérébrales : syndrome de la selle turcique vide et autres anomalies osseuses, notamment angle du clivus raide, anomalies de la glande pituitaire avec le plus souvent hypoplasie de l’hypophyse entraînant un retard de croissance par déficit en hormone de croissance et enfin anomalie de la dure-mère (méningiome);
cardiaques et anomalies des gros troncs aortiques.
L’aniridie est une pathologie sévère du développement de l’œil; elle ne se limite pas à la simple absence de l’iris. L’ensemble de l’œil est anormal; il existe une cataracte le plus souvent antérieure, limitant peu la vision. Le pronostic fonctionnel est lié à l’hypoplasie fovéale constante, à l’insuffisance limbique responsable de néovascularisation cornéenne périphérique et d’opacifications de la cornée, et au glaucome (Fig. 11-6). L’aniridie est la conséquence d’une atteinte du gène PAX6, selon une transmission dominante autosomique [15]. Cette atteinte peut être :
soit génétique isolée, conséquence de mutations variables de ce gène, pouvant entraîner, un phénotype d’aniridie et aussi d’autres types de malformations oculaires (absence de corrélation génotype-phénotype) [16];
soit la conséquence d’une microdélétion en 11 p 13, où se situe le gène PAX6. La réalisation d’un caryotype en haute résolution est indispensable car les gènes adjacents à PAX6 peuvent être aussi emportés dans la délétion.
Il est nécessaire de rechercher un syndrome de gènes contigus appelé syndrome WAGR (Wilms tumor, Aniridia, Genital anomalies, mental Retardation). En effet, la tumeur de Wilms est un néphroblastome et il est important de dépister ce cancer de façon précoce par des échographies abdominales itératives; de même, il peut exister des gonadoblastomes à rechercher. Le glaucome de l’aniridie est tardif, survient dans les premières années de vie, et n’entraîne ni buphtalmie ni œdème de cornée à l’inverse du glaucome congénital primitif. Il est de diagnostic difficile car nécessairement réalisé lors d’examens sous anesthésie générale (car le nystagmus rend difficile la mesure de la tension oculaire en consultation).
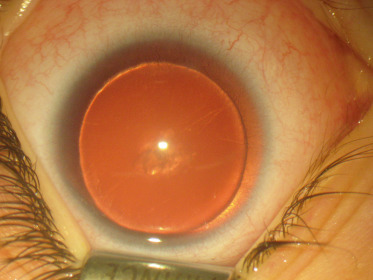
Fig. 11-6 Aniridie.
Voir chapitre 12.
[1] Johnston MC, Noden DM, Hazelton RD, et al. Origins of avian ocular and periocular tissues Exp Eye Res 1979 ; 29 : 27-43
[2] Hoskins HD, Shaffer RN, Hetherington J Anatomical classification of the developmental glaucomas Arch Ophthalmol 1984 ; 102 : 1331-1336
[3] Nischal KK Genetics of congenital corneal opacification – impact on diagnosis and treatment Cornea 2015 ; 34 (Suppl 10) : S24-S34
[4] Patel SP, Parker MD SLC4A11 and the pathophysiology of congenital hereditary endothelial dystrophy Biomed Res Int 2015 ; 2015 : 475392
[5] Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies – edition 2 Cornea 2015 ; 34 : 117-159Erratum in. Cornea 2015 ; 34 : e32
[6] Michelle J, Kim Ricardo F, et al. Aldave. Posterior amorphous corneal dystrophy is associated with a deletion of small leucine-rich proteoglycans on chromosome 12 PLoS One 2014 ; 9 : e95037
[7] de Almeida JC, Reis DF, Llerena J, et al. Short stature, brachydactyly, and Peters’ anomaly (Peters’-plus syndrome) : confirmation of autosomal recessive inheritance J Med Genet 1991 ; 28 : 277-279
[8] Lesnik Oberstein SA, Kriek M, White SJ, et al. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase Am J Hum Genet 2006 ; 79 : 562-566
[9] Bhandari R, Ferri S, Whittaker B, Liu M, et al. Peters anomaly : review of litterature vCornea 2011 ; 30 : 939-944
[10] Shigeyasu C, Yamada M, Mizuno Y, et al. Clinical features of anterior segment dysgenesis associated with congenital corneal opacities Cornea 2012 ; 31 : 293-298
[11] Schramm C, Rohrbach JM, Reinert S, et al. Amniotic bands as a cause of congenital anterior staphyloma Graefes Arch Clin Exp Ophthalmol 2013 ; 251 : 959-965
[12] Al Judaibi R, Abu-Amero KK, Morales J, et al. Mutations of the CYP1B1 gene in congenital anterior staphylomas Clin Ophthalmol 2014 ; 8 : 445-448
[13] Tümer Z, Bach-Holm D Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations Eur J Hum Genet 2009 ; 17 : 1527-1539
[14] Amendt BA, Semina EV, Alward WL Rieger syndrome : a clinical, molecular, and biochemical analysis Cell Mol Life Sci 2000 ; 57 : 1652-1666
[15] Kokotas H, Petersen MB Clinical and molecular aspects of aniridia Clin Genet 2010 ; 77 : 409-420
[16] Hingorani M, Williamson KA, Moore AT, Van Heyningen V Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations Invest Ophthalmol Vis Sci 2009 ; 50 : 2581-2590
E. Bui Quoc
Les opacités congénitales acquises de la cornée sont multiples. Ici encore, on retrouve un paradoxe entre le terme « congénital » (on naît avec la pathologie) et le terme « acquis » qui sous-entend un délai de survenue.
Premier élément de réflexion, l’anomalie peut être acquise in utero. Second élément, une anomalie innée/malformative peut ne se manifester que tardivement. Par exemple, dans le glaucome congénital tardif dans lequel l’anomalie génétique est constitutive, l’opacité de cornée révèle la pathologie ne survenant que plus tard.
Nous évoquons ici les causes secondaires et acquises des opacités congénitales de la cornée, selon la classification de Nischal [1, 2]. Il ne s’agit pas de dysgénésies du segment antérieur (anomalie innée car anomalie génétique présente même si elle n’est pas caractérisée). Le diagnostic « doit » ou « peut » être un diagnostic d’élimination selon les antécédents et l’interrogatoire, l’évolutivité de la pathologie et l’analyse sémiologique précise.
Les causes sont : infectieuses, traumatiques, métaboliques ou autres.
Le caractère infectieux d’une opacité de cornée est orienté par le contexte clinique et les signes d’examen. L’opacité de cornée peut être congénitale en cas de rubéole contractée pendant la grossesse, mais de nombreuses infections peuvent être en cause, avec les classiques causes à rechercher : toxoplasmose, syphilis/virus de la varicelle et du zona/parvovirus B19 (autres : rubéole, cytomégalovirus, herpès, chorioméningite lymphocytaire virale ou virus du Nil occidental [3]). L’infection cornéenne avec abcès peut survenir après la naissance et est le contexte révélateur. À titre d’exemple, le cas d’un nourrisson qui présente une paralysie faciale unilatérale congénitale avec ulcère d’exposition se compliquant d’un abcès (Fig. 11-7).
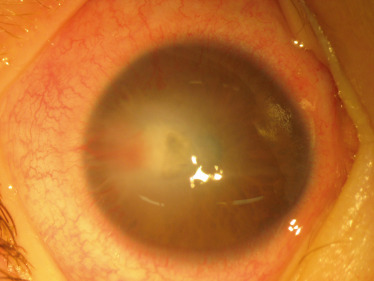
Fig. 11-7 Ulcère d’exposition secondaire à une paralysie faciale unilatérale congénitale se compliquant d’un abcès.
Là encore, le traumatisme est le plus souvent évident. En postnatal, les conditions traumatiques sont variées : griffure cornéenne se compliquant de kératoconjonctivite et d’abcès, lésions par un objet comme un jouet, etc. En périnatal, une opacité de cornée peut survenir après un accouchement par forceps, et outre la question étiologique de l’opacité présente (l’opacité n’étant pas forcément imputable au forceps), peut se poser une question médico-légale car parfois une procédure peut être en cours. L’application de forceps, outre le traumatisme orbitaire possible, peut entraîner une opacification de la cornée par deux mécanismes :
ruptures de la Descemet, volontiers verticales, à la différence des ruptures de Descemet horizontales du glaucome congénital; elles sont invisibles à l’œil nu et nécessitent un examen avec une lampe à fente pour être éventuellement diagnostiquées. La question pratique est la possible conséquence réfractive avec anisométropie précoce par astigmatisme induit;
hyphéma pouvant se compliquer d’hypertonie oculaire, d’œdème de cornée, d’hématocornée. Dans ce cas, une intervention chirurgicale précoce peut être requise : lavage de la chambre antérieure, chirurgie filtrante.
De nombreuses maladies métaboliques peuvent entraîner une anomalie de la transparence des yeux. Les opacités de cornée sont ici rarement congénitales et plutôt d’apparition tardive. Dans ce groupe, on peut retrouver les mucolipidoses et l’ensemble des mucopolysaccharridoses : maladie de Hurler (MPS1H), maladie de Scheie (MPS1S), maladie de Hurler-Scheie (MPS1H/S), maladie de Hunter (MPS2), maladie de San Filippo (MPS3), maladie de Morquio (MPS4), maladie de Maroteaux-Lamy (MPS6), maladie de Sly (MPS7), maladie de Natowicz (MPS 9).
On peut retrouver également la cystinose.
Des maladies rares peuvent entraîner des opacités de cornée, comme ce cas d’histiocytose, avec opacification subtotale de la cornée apparue pendant la première année de vie (Fig. 11-8). Parfois la cause demeure inconnue, comme dans ce cas d’opacité unilatérale superficielle épithéliale et sous-épithéliale (Fig. 11-9). Celle-ci n’est pas liée à une dysgénésie du segment antérieur et ne présente pas de synéchies iridocornéennes pouvant être caractéristiques d’une anomalie de Peters. L’UBM ou l’OCT du segment antérieur est un outil précieux pour l’analyse sémiologique du segment antérieur [4].
Les opacités congénitales secondaires acquises de la cornée représentent un groupe de pathologies variées d’origine traumatique, infectieuse, métabolique, parfois « idiopathique » .
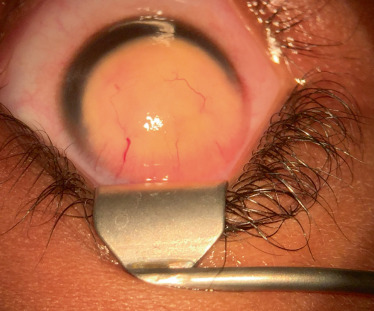
Fig. 11-8 Histiocytose, avec opacification subtotale de la cornée apparue pendant la première année de vie.
(Source : remerciements au Dr L. Vera.)
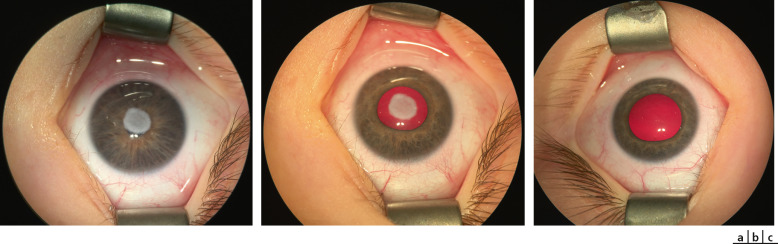
Fig. 11-9 Opacité unilatérale superficielle épithéliale et sous-épithéliale.
a. Opacité congénitale idiopathique. b. OEil après dilatation. c. OEil controlatéral.
[1] Nischal KK Genetics of congenital comeal opacification – impact on diagnosis and treatment Cornea 2015 ; 34 : S24-S34
[2] Kurilec JM, Zaidman GW Incidence of Peters anomaly and congenital corneal opacities interfering with vision in the United States Cornea 2014 ; 33 : 848-850
[3] Mets MB, Chhabra MS Eye manifestations of intrauterine infections and their impact on childhood blindness Surv Ophthalmol 2008 ; 53 : 95-111
[4] Majander AS, Lindahl PM, Vasara LK, Krootila K Anterior segment optical coherence tomography in congenital corneal opacities Ophthalmology 2012 ; 119 : 2450-2457
E. Bui Quoc
Les traitements des dysgénésies du segment antérieur et des opacités congénitales de la cornée sont difficiles et longs. Comme le soulignent Medsinge et Nischal en ce qui concerne la greffe de cornée chez l’enfant [1], le problème est une question de choix et demeure un casse-tête insoluble. La question de la greffe de cornée n’est pas la seule, le glaucome peut être sévère et cécitant, et il est fondamental de prendre en compte la problématique de la réhabilitation visuelle et de l’amblyopie.
La question de la maturation de la fonction visuelle au cours de la période sensible du développement visuel est à souligner, avec une différence majeure selon que l’anomalie est unilatérale ou bilatérale :
dans un cas unilatéral (exemple de l’anomalie de Peters de type 1 avec opacification cécitante de la cornée), le dilemme est le suivant : même si on rétablit la transparence cornéenne (greffe ou kératoprothèse), le résultat fonctionnel dépendra de la précocité du traitement chirurgical;
dans un cas bilatéral, en l’absence de tout traitement précoce, le cerveau visuel se développera de façon anormale; la cécité bilatérale précoce (quelle que soit la cause oculaire) entraîne une réorganisation corticale majeure qui compromet le résultat fonctionnel si le traitement n’est pas entrepris tôt. C’est-à-dire que, par exemple, dans un cas d’anomalie de Peters de type 1 bilatéral, si la transparence de la cornée n’est pas rétablie précocement, l’amblyopie sera profonde et bilatérale, même après traitement chirurgical au résultat anatomique satisfaisant.
Plusieurs questions préalables doivent se poser en cas de dysgénésie du segment antérieur et/ou d’opacité congénitale de la cornée : quel est le diagnostic précis? La pathologie est-elle innée ou acquise, évolutive ou stable? Où se situe l’opacité si elle existe : épithéliale, stromale, endothéliale? Quels sont les moyens thérapeutiques (chirurgie filtrante du glaucome – laquelle? –, greffe de cornée transfixiante, lamellaire, endothéliale, autres thérapeutiques, etc.)?
Comme nous l’avons souligné de façon itérative, l’enjeu de la prise en charge d’un enfant en ophtalmologie pédiatrique doit être de lui permettre une vie la plus normale possible, et d’obtenir une vision de plus de 5/10. Est-ce une chimère dans la prise en charge des opacités congénitales de cornée et des dysgénésies du segment antérieur? Reprenons l’exemple de l’anomalie de Peters de type 1 : les objectifs demeurent bien plus modestes qu’en cas de cataracte congénitale, uni- ou bilatérale, en particulier car la greffe de cornée chez l’enfant n’a pas la même facilité (ou plutôt est encore plus difficile) que la chirurgie de la cataracte, et la survie du greffon (ou de la kératoprothèse) à moyen terme est une question extrêmement problématique. Mais, encore une fois, il faut raisonner et ne pas a priori être catégorique en affirmant : « Il faut/il ne faut pas greffer une cornée chez un enfant. »
La chirurgie du glaucome congénital isolé est difficile, avec diverses techniques proposées : goniotomie, trabéculotomie, trabéculectomie, sclérectomie profonde.
La goniotomie serait efficace dans 91 % des cas selon François [2], dans 88 % des cas selon Broughton et Parks [3], dans 86 % des cas selon Shaffer et Hoskins [4]; il est à noter dans cette dernière série 46 % d’amblyopie.
La trabéculotomie est une alternative à la goniotomie. Meyer et al. retrouvent, dans une série de 29 yeux de 22 patients traités par cette procédure (renouvelée au besoin avec un nombre moyen de procédures de 1,3), un succès défini comme un tonus oculaire de moins de 21 mmHg dans 79,5 % des yeux à la fin du suivi, celui-ci étant en moyenne de 24,7 ± 17,9 mois [5].
La trabéculectomie avec iridectomie est une alternative efficace mais avec risque d’hypotonie, ce qui nous fait préférer la sclérectomie profonde comme Denis et al. qui retrouvent, sur 18 yeux opérés par sclérectomie avec application de 5-fluorouracile, un succès total défini comme un tonus oculaire inférieur à 16 mmHg à la fin du suivi sans autre thérapeutique dans 56 % des cas (dans 89 % des cas, un succès est obtenu avec adjonction éventuelle d’une thérapeutique complémentaire) [6].
Dans le cas des glaucomes dysgénésiques et/ou secondaires, la nature de l’obstacle à la filtration doit être analysée et la chirurgie perforante semble à privilégier, comme dans l’anomalie d’Axenfeld-Rieger et comme le montre cette publication indienne [7] présentant une série de 44 yeux de 24 patients opérés par 6 trabéculectomies ou 38 trabéculotomies-trabéculectomies (lorsque le canal de Schlemm pouvait être identifié). La réduction tensionnelle est de 45,14 % en moyenne, avec un tonus préopératoire de 27,07 ± 4,88 mmHg, contre 14,88 ± 3,62 mmHg en postopératoire (p < 0,0001).
Le glaucome doit donc être diagnostiqué avec attention dans toutes les dysgénésies du segment antérieur et traité chirurgicalement le plus souvent.

Fig. 11-10 Anomalie de Peters de type 1 et cataracte.
OEil droit : aspect préopératoire (a) et postopératoire à 7 mois (c). OEil gauche : aspect préopératoire (b) et postopératoire à 13 mois (d).
Les greffes de cornée peuvent être transfixiantes, lamellaires, endothéliales. Le choix doit dépendre de la localisation de l’opacité et des possibilités techniques.
Schématiquement, le greffe de cornée transfixiante est la seule alternative dans l’anomalie de Peters avec opacité de pleine épaisseur épithéliale, stromale, endothéliale, ou dans la sclérocornée. Une problématique majeure est la question de la survie du greffon à long terme [1] ayant fait développer dans ces indications les kératoprothèses [8]. La greffe de cornée lamellaire est possible par exemple pour une opacité superficielle post-traumatique ou une pathologie métabolique de surcharge n’affectant pas l’endothélium [9]. La greffe de cornée endothéliale est une alternative dans les dystrophies cornéennes congénitales héréditaires [10, 11].
La greffe de cornée transfixiante et la kératoprothèse ont un pronostic anatomique et fonctionnel conditionné :
au plan anatomique du fait du risque de rejet, des complications per- et postopératoires [12, 13, 14, 15];
au plan fonctionnel par l’amblyopie possible dans les formes unilatérales, mais aussi dans les formes bilatérales du fait d’une possible anisométropie et/ou d’un résultat anatomique asymétrique [16, 17].
Nos illustrations montrent deux cas d’anomalie de Peters bilatéral avec résultat postopératoire satisfaisant à moyen terme (Fig. 11-10 et Fig. 11-11).
La greffe de cornée transfixiante chez l’enfant pour une dysgénésie du segment antérieur ou une opacité de cornée précoce doit être précoce, dans la première année de vie, pour préserver un pronostic fonctionnel relatif. La greffe en elle-même est techniquement difficile, peut s’accompagner de chirurgie de la cataracte dans le même temps, peut être faite sur un œil relativement microphtalme. La greffe doit être de petite taille (< 7 mm avant 1 an si possible) pour réduire le risque de rejet et anticiper des greffes ultérieures. Après la greffe, des examens sous anesthésie générale itératifs sont requis, quasiment tous les mois pendant 6 mois, pour retirer progressivement les fils. Une surveillance ultérieure avec en particulier recherche de signes de rejet et recherche de glaucome (associé, iatrogène par inflammation ou secondairement au traitement corticoïde) doit être extrêmement régulière et attentive. L’introduction initiale d’un traitement immunosuppresseur local (ciclosporine collyre à 2 % ) est indispensable pour réduire le risque de rejet.
L’infection précoce ou tardive est possible et ses conséquences sont lourdes (abcès à Candida par exemple : Fig. 11-12).
Le greffon n’est pas fonctionnel immédiatement (œdème du greffon, déformation et astigmatisme irrégulier évolutif avec ablation progressive des fils de cornée). C’est pourquoi le pronostic peut être compromis par ce délai entre naissance et greffe (6 mois par exemple) et le délai entre greffe et fonctionnalité du greffon (6 à 12 mois post-greffe) du fait de l’altération de la maturation visuelle au cours de cette période « très » critique du développement visuel. C’est pourquoi il a été proposé des kératoprothèses, à la fonctionnalité optique immédiate, même si les complications de ce dispositif sont non négligeables : membrane rétroprothétique, impossibilité de faire une réfraction objective à l’autoréfractomètre, évaluation du tonus oculaire et examen du fond d’œil difficiles (Fig. 11-13). Notez cependant que dans le cas de ce patient illustré dans la Fig. 11-13, à l’âge de 5 ans, une endophtalmie endogène a conduit à l’énucléation de l’œil…
La kératoprothèse est une procédure proposée par certains en première intention non seulement dans les anomalies de Peters, mais aussi dans la sclérocornée, certes dont le pronostic de la greffe transfixiante est moins bon que dans l’anomalie de Peters, mais pouvant donner un résultat anatomique satisfaisant à moyen terme (Fig. 11-14).
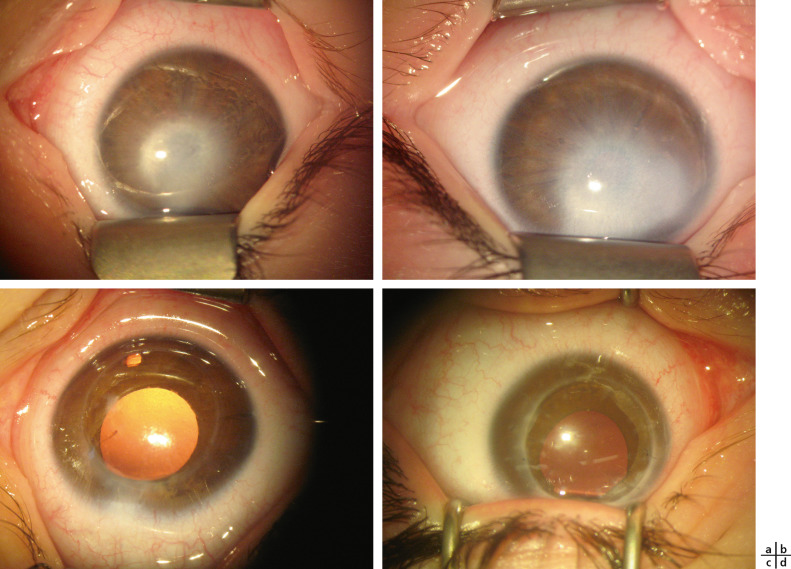
Fig. 11-11 Anomalie de Peters de type 1
OEil droit : aspect préopératoire – opacité (a) et postopératoire à 23 mois (c). OEil gauche : aspect préopératoire (b) et postopératoire à 27 mois (d).
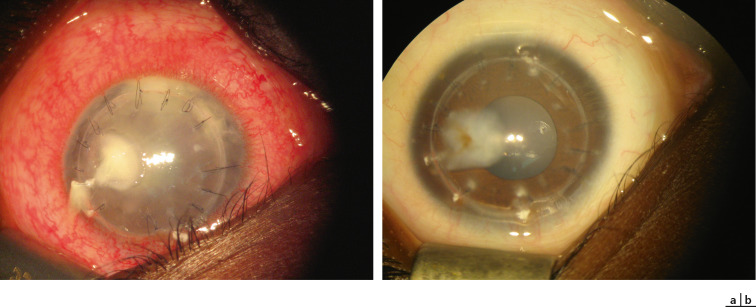
Fig. 11-12 Abcès de cornée à Candida albicans.
a. Post-greffe chez un enfant à 6 mois. b. Cicatrisation opaque à 33 mois de la greffe.
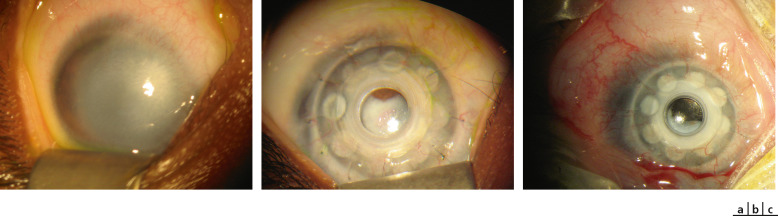
Fig. 11-13 Anomalie de Peters unilatérale traitée par kératoprothèse outre-Atlantique, une greffe unilatérale n’ayant pas été proposée en France.
a. Anomalie de Peters de type 1 (oeil gauche aspect préopératoire). b. Kératoprothèse compliquée de membrane rétroprothétique. c. Aspect après ablation de la membrane.
Après une greffe de cornée transfixiante pour opacification congénitale de cornée, avant l’âge de 3 à 4 ans (c’est-à-dire essentiellement pour des pathologies malformatives dysgénésiques, comme l’anomalie de Peters), on ne devrait pas parler de traitement de l’amblyopie unilatérale, mais de traitement de l’amblyopie bilatérale. En effet, nous le détaillerons plus loin, la greffe de cornée ne devrait être proposée dans ce cas plutôt en cas d’anomalie bilatérale (sauf exceptions).
Le raisonnement est en effet différent si on le compare à celui de la cataracte congénitale. L’objectif en cas de cataracte congénitale unilatérale est d’obtenir une fonction visuelle à 6 ans supérieure à 5/10, grâce à une chirurgie précoce avec implantation et rééducation acharnée de l’amblyopie (soit, pour résumer, occlusion de la moitié aux trois quarts du temps d’éveil jusqu’à 6 ans); l’objectif est le même en cas de cataracte bilatérale, avec occlusion différente éventuellement alternée. Rappelons qu’il y a 30 ans, la chirurgie de la cataracte congénitale unilatérale se discutait car le pronostic fonctionnel était médiocre en l’absence d’implantation; c’est probablement le même raisonnement qu’il faut avoir en cas d’anomalie de Peters ou de sclérocornée unilatérale, avec en plus la problématique de la difficulté de pérenniser un bon résultat anatomique initial (rejet ou œdème du greffon par insuffisance endothéliale à moyen/long terme). Certes, il est toujours vrai que dans la cataracte unilatérale précoce, l’œil opéré est un œil « de secours » s’il survenait un problème sur l’autre œil, car le plus souvent, l’œil non opéré demeure dominant avec fixation monoculaire et absence de vision binoculaire. Dans les opacités congénitales unilatérales de cornée qui nécessiteraient de rétablir la transparence cornéenne par une greffe transfixiante, le pronostic anatomique et fonctionnel est compromis à long terme.
La question de la réhabilitation visuelle après greffe de cornée (bilatérale) requiert donc de respecter les principes de base du traitement de l’amblyopie : évaluations répétées de la réfraction (objective et subjective), occlusion alternée pour obtenir une alternance de fixation, occlusion unilatérale en cas d’amblyopie unilatérale.
Enfin, la vision peut être limitée à quelques dixièmes, avec nystagmus sensoriel, ce qui peut nécessiter l’accompagnement de l’enfant et des parents dans un parcours scolaire normal avec aides ou dans un circuit éducatif spécialisé.
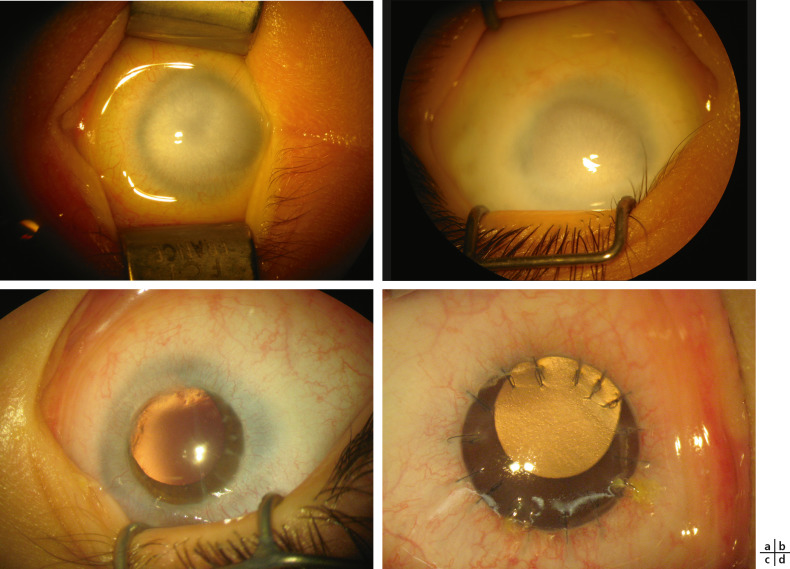
Fig. 11-14 Sclérocornée bilatérale : aspect postopératoire à court et moyen terme.
OEil droit : aspect préopératoire (a) et postopératoire à 34 mois (c). OEil gauche : aspect préopératoire (b) et postopératoire à 2 mois (d).
Les procédures de greffes lamellaires et de greffes endothéliales sont proposées le plus souvent après la fin de la période sensible du développement visuel, dans des pathologies autres que les dysgénésies sévères du segment antérieur.
La greffe lamellaire peut être proposée dans les pathologies métaboliques, par exemple la mucopolysaccharidose en l’absence d’anomalie endothéliale. Par exemple, Sati et al. [9] montrent un cas de kératoplastie lamellaire profonde dans un cas de syndrome de Hurler-Scheie, chez un enfant de 12 ans, avec un bon résultat anatomique (greffon clair) et fonctionnel à 1 an : acuité visuelle de 20/50 avec + 1,00 (170° – 0,75), et un décompte de cellules endothélial de 2473,4 cellules/mm2.
Ashar et al. [18] comparent la procédure de greffe endothéliale à la kératoplastie transfixiante dans les CHED et montrent dans une série de patients opérés à un âge moyen de 6,6 ± 2,19 ans que le résultat anatomique est similaire, avec une réhabilitation visuelle plus rapide dans le groupe des greffes endothéliales.
La même équipe [19] présente une série de 5 yeux de patients traités par kératoplastie endothéliale (Descemet’s stripping endothelial keratoplasty ou DSEK) chez des patients présentant une CHED et opérés à un âge moyen de 7,8 ans (5-12 ans). À 1 an, les cornées sont claires avec normalisation de l’épaisseur cornéenne.
Greffes de cornée transfixiantes (et kératoprothèses) sont indiquées chez des enfants beaucoup plus jeunes, pour des pathologies différentes (opacités dysgénésiques).
Notre expérience personnelle sur le traitement de 17 yeux de 10 patients atteints de dysgénésie sévère du segment antérieur (11 anomalies de Peters de type 1 et 6 sclérocornées bilatérales chez 7 patients et unilatérales chez 3 patients) montre que, sur une période de 4 ans, 13 yeux ont été traités par greffe de cornée et 1 œil par kératoprothèse. Le suivi moyen était de 23,2 mois (déviation standard ou DS : 13,95 mois) allant de 3 mois à 43 mois, le temps médian de suivi était de 24 mois. Sur les 14 yeux opérés, 12 yeux ont eu une seule greffe de cornée, 1 œil a bénéficié de deux greffes de cornée, 1 œil a eu une kératoprothèse. L’âge moyen de la première chirurgie était de 8,6 mois. L’âge moyen de la chirurgie était de 12,57 mois (2-29 mois; DS : 9,46 mois), l’âge médian de la première chirurgie de chaque œil opéré était de 9 mois. Huit yeux atteints d’anomalie de Peters ont donc été traités par kératoplastie transfixiante avec un résultat satisfaisant dans 6 cas; 2 yeux (du même patient) ont présenté un rejet sévère après abcès cornéen (observance thérapeutique aléatoire); 1 œil a présenté un abcès fongique résolutif sous traitement. Une forme unilatérale a été traitée par kératoprothèse (pas par notre équipe) avec un résultat anatomique et fonctionnel médiocre. Parmi les patients étudiés, nous avions 6 cas de sclérocornée : 2 yeux d’un même patient opérés avec un bon résultat, 2 yeux d’un autre patient avec résultat médiocre pour un œil et rejet sur l’autre œil; 1 cas unilatéral non opéré, un dernier cas compliqué d’ulcère chronique puis d’abcès avec opacification du greffon. Cette brève analyse montre que le pronostic des greffes en cas d’anomalie de Peters de type 1 semble meilleur qu’en cas de sclérocornée. Nos résultats semblent comparables à la littérature, même si notre effectif est réduit : 75 % de résultats satisfaisants dans l’anomalie de Peters de type 1, 40 à 60 % dans la sclérocornée (2 yeux sur les 5 avec un résultat satisfaisant et 1 œil avec un résultat moyen).
Yang [12], dans une série de 47 patients atteints d’anomalies de Peters (72 yeux greffés) avec un âge médian de la première chirurgie à 4,4 mois, retrouvait un taux de greffons clairs de : 56 % à 6 mois, 49 % à 1 an, 44 % à 3 ans et 35 % à 10 ans. Frueh et Brown retrouvaient 74 % de survie du greffon à 1 an sur une série 58 yeux atteints d’opacités congénitales (27 slérocornées, 12 anomalies de Peters, 12 sclérocornées partielles, 2 opacités cornéennes sur glaucome congénital) [13]. Le taux de greffons clairs passait à 58 % à 2 ans.
Patel, dans une étude sur une série de 58 yeux de 52 enfants atteints d’opacités cornéennes congénitales (anomalie de Peters, CHED, autres opacités congénitales non étiquetées) ou acquises (traumatiques et non traumatiques), retrouve un taux de greffons clairs de 78 % à 1 an sur les 9 enfants atteints d’opacité congénitale, de 85 % dans les 43 cas d’atteintes non traumatiques acquises (kératite virale, kératocône) et de 100 % à 1 an dans les 6 cas d’atteintes traumatiques [14].
Comer présente lui une étude concernant 16 yeux de 11 enfants dont 5 ont bénéficié d’une kératoplastie bilatérale, avec au total 26 kératoplasties réalisées entre l’âge de 2 à 56 semaines. Sur 16 yeux (10 greffes réalisées sur des yeux déjà greffés), il retrouve une évolution favorable à 1 an du point de vue anatomique dans 61 % des cas, c’est-à-dire 10 yeux sur 16, et retrouve une survie des greffons comprise entre 3 et 137 mois [20].
La problématique du résultat anatomique n’est pas la seule à prendre en compte. L’autre problème une fois la transparence cornéenne rétablie est celui de la fonction visuelle et de l’amblyopie, liées à l’amétropie et l’anisométropie. Une étude sur les résultats fonctionnels à long terme a été réalisée par Zaidman concernant 24 enfants atteints d’anomalie de Peters de type 1 greffés, dont 16 avec une atteinte unilatérale et 8 avec une atteinte bilatérale (au total 30 yeux greffés) [17]. Sur les 24 yeux des enfants évalués à l’âge verbal, l’acuité visuelle postopératoire est de 20/20 à 20/50 dans 7 yeux, de 20/50 à 20/100 dans 9 yeux, inférieure ou égale à 20/100 dans 11 yeux, pour une durée de suivi moyenne de 78,9 mois. Najjar présente également une série de 25 patients atteints d’anomalie de Peters bilatérale dont 20 ont bénéficié de kératoplastie transfixiante avec au total 34 yeux opérés [16]. La durée moyenne du suivi était de 5 ans. Dans 40 % des cas, l’acuité visuelle à la fin du suivi était supérieure à 20/400, dans 36 % inférieure ou égale à 20/400. Dans 9 % des cas, il n’y avait pas de perception lumineuse.
Dans les cas d’atteinte bilatérale de la transparence cornéenne, une chirurgie devrait être proposée car le pronostic est quasiment nul en l’absence d’intervention. ll est cependant difficile d’établir des règles précises compte tenu des différents degrés d’atteinte cornéenne. Le cas des atteintes unilatérales pose quant à lui le problème du rapport bénéfice/risque. Faut-il greffer les atteintes unilatérales connaissant les risques de rejet, d’infections et d’amblyopie?
C’est cette interrogation que souligne Basdekidou et al. [21] après l’analyse de leur série de 14 patients présentant une anomalie de Peters unilatérale greffée (greffe transfixiante) sur une période de 10 ans. L’âge moyen de chirurgie était de 9 mois; le suivi moyen était de 30 mois. Si le résultat anatomique est satisfaisant dans 78,6 % des cas (11 yeux) de la série, le résultat fonctionnel est sujet à caution : acuité visuelle mesurable à 20/50 dans 1 cas, 20/63 dans 1 cas, et 20/2000 dans 1 cas; chez les 8 autres patients à l’âge préverbal, la fixation est possible dans tous les cas.
Une série coréenne présente les résultats de la kératoplastie transfixiante pour anomalie de Peters ou sclérocornée dans 20 yeux de 18 patients, opérés avant l’âge de 5 ans (8 cas d’anomalie de Peters, 10 cas de sclérocornée). Au cours du suivi de 92,7 ± 10 mois, on note un taux de survie du greffon de 50 % (65 % à 6 mois et 50 % à 1, 2 et 5 ans). Le temps de survie est significativement meilleur en cas d’anomalie de Peters qu’en cas de sclérocornée (135,6 17,9 versus 36,4 ± 16,1 mois, p = 0,014; taux de survie 87,5 % versus 25,0 % , p = 0,02) [22].
Ces résultats parfois décevants conduisent à discuter l’option d’une kératoprothèse de première intention ou après rejet d’une première greffe. Kosker et al. [23] présentent une série de 37 yeux de 37 patients traités avec une kératoprothèse Optical BKPro® (kératoprothèse de Boston; voir Fig. 11-13c) pour rejet d’une première kératoplastie transfixiante dans 28 cas, et thérapeutique primaire dans 9 cas. L’âge de la greffe était de 31,7 ± 21 mois (12-78 mois). Le taux de complication est élevé (glaucome et membrane rétroprothétique dans 43 % des cas). L’œil controlatéral était sain (acuité visuelle ≥ 20/40); le résultat fonctionnel retrouve une amélioration de l’acuité visuelle : postopératoire (= 1,0 ± 0,8 logMAR; médiane, 1,0 logMAR) versus préopératoire (= 1,8 ± 0,2 logMAR; médiane, 1,8 logMAR, p < 0,001).
En cas d’opacité acquise, le choix thérapeutique va dépendre de l’étiologie. Une atteinte traumatique avec opacification de l’ensemble de la cornée requiert une kératoplastie transfixiante, de pronostic fonctionnel réservé selon la date de survenue du traumatisme.
Une pathologie de surcharge peut requérir une greffe lamellaire avec de bons résultats chez le sujet jeune [9] ou transfixiante avec des résultats satisfaisants dans cette dernière option chez des patients plus âgés, comme le montre cette étude américaine [24], concernant 8 yeux de 5 patients présentant une MPS avec une greffe entre 11,7 et 65,3 ans. À cet âge ne se pose pas le problème de l’amblyopie et les résultats fonctionnels à l’issue du suivi (4,9 ans en moyenne; 1 à 11 ans) montrent une acuité visuelle de 0,32 (0,16) logMAR en postopératoire contre 0,90 (0,38) logMAR en préopératoire.
Les dysgénésies du segment antérieur sont des affections rares pouvant avoir des conséquences fonctionnelles graves du fait du glaucome ou des opacités de cornée. Les opacités congénitales de cornée peuvent exister chez l’enfant indépendamment d’une dysgénésie du segment antérieur, dans le cadre de pathologies endothéliales, ou de pathologie acquise, métabolique, traumatique, infectieuse.
En pleine période sensible du développement visuel, ces troubles sévères de la transparence cornéenne posent particulièrement le problème d’une malvoyance possible, et le problème de l’amblyopie sévère, même après rétablissement d’une transparence cornéenne utile dans les formes unilatérales. Dans les cas de formes bilatérales, l’amblyopie bilatérale est une problématique prégnante. Une anomalie de transparence de la cornée induit une amblyopie à la fois organique et fonctionnelle. C’est pourquoi la kératoplastie peut être nécessaire rapidement, même si cela reste une thérapeutique dont le résultat fonctionnel final est aléatoire, la greffe étant « fonctionnelle » au bout de 6 mois environ après ablation des fils. Cette période de latence constitue un risque supplémentaire dans l’apparition d’une amblyopie. Cette période est en théorie absente lorsqu’il s’agit d’une mise en place d’une kératoprothèse (fonctionnelle immédiatement), ce qui constitue la justification de cette procédure pour les équipes préconisant cette thérapeutique, non dénué de risques cependant. La kératoplastie transfixiante est un traitement lourd non sans risque. Avant même que se pose la problématique de l’efficacité optique du greffon, l’enfant est exposé à de nombreuses complications postopératoires graves qui sont en général plus fréquentes chez l’enfant que chez l’adulte et qui débouchent par conséquent sur des résultats globalement moins bons chez l’enfant que chez l’adulte. Le rejet et les infections sont les principales causes d’échec de cette chirurgie.
Il faut privilégier si possible les kératoplasties lamellaires ou endothéliales lorsque la pathologie l’autorise.
La greffe de cornée en cas d’anomalie de Peters ou de sclérocornée est une solution thérapeutique dont le pronostic est plus réservé en ce qui concerne la sclérocornée. La prise en charge doit être précoce. Cependant il s’agit d’une solution thérapeutique lourde en termes de suivi avec un risque d’échec non négligeable lié au risque de perte de transparence du greffon par rejet ou infection, comme nous avons pu le constater dans notre série. Par ailleurs, le greffon a sa propre durée de vie et l’insuffisance endothéliale à moyen ou long terme (par simple « vieillissement » du greffon) est une cause de perte de transparence possible.
Alors faut-il greffer? Faut-il privilégier la kératoprothèse? Quel que soit le choix d’une greffe transfixiante ou d’une kératoprothèse, l’indication est positive dans les cas bilatéraux, mais discutable dans les cas unilatéraux. La sévérité de la dysgénésie, le caractère uni- ou bilatéral sont des critères qui doivent entrer en ligne de compte pour envisager le projet thérapeutique, invasif ou non, avec le souci, quelle que soit la situation, d’un accompagnement adapté en cas de malvoyance.
Concernant le glaucome, il est redoutable dans les dysgénésies du segment antérieur et doit être diagnostiqué, dépisté et traité avec attention lorsqu’il survient.
[1] Medsinge A, Nischal KK Paediatric keratoplasty : choices and conundrums Br J Ophthalmol 2013 ; 97 : 1225-1227
[2] Francois J, Van Oye R, Mendoza A, de Sutter E La goniotomie dans le glaucome congénital J Fr Ophtalmol 1982 ; 5 : 661-664
[3] Broughton WL, Parks MM An analysis of treatment of congenital glaucoma by goniotomy Am J Ophthalmol 1981 ; 91 : 566-572
[4] Shaffer RN, Hoskins HD Montgomery lecture. Goniotomy in the treatment of isolated trabeculodysgenesis (primary congenital [infantile] developmental glaucoma) Trans Ophthalmol Soc U K 1983 ; 103 : 581-585
[5] Meyer G, Schwenn O, Pfeiffer N, Grehn F Trabeculotomy in congenital glaucoma Graefes Arch Clin Exp Ophthalmol 2000 ; 238 : 207-213
[6] Denis D, Pommier S, Coste R, et al. Sclérectomie profonde dans le glaucome congénital J Fr Ophtalmol 2008 ; 31 : 173-179
[7] Mandal AK, Pehere N Early-onset glaucoma in Axenfeld-Rieger anomaly : long-term surgical results and visual outcome Eye (Lond) 2016 ; 30 : 936-942
[8] Aquavella JV Pediatric keratoprosthesis : a new surgical approach Ann Ophthalmol (Skokie) 2008 ; 40 : 64-67
[9] Sati A, Ramappa M, Chaurasia S, Prasad SM Deep anterior lamellar keratoplasty in case of Hurler-Scheie syndrome BMJ Case Rep 2014 ; 2014 : pii : bcr2013202730.
[10] Anwar HM, El-Danasoury A Endothelial keratoplasty in children Curr Opin Ophthalmol 2014 ; 25 : 340-346
[11] Ramappa M, Ashar J, Vaddavalli PK, et al. Endothelial keratoplasty in children : surgical challenges and early outcomes Br J Ophthalmol 2012 ; 96 : 1149-1151
[12] Yang LL, Lambert SR, Lynn MJ, Stulting RD Long-term results of corneal graft survival in infants and children with peters anomaly Ophthalmology 1999 ; 106 : 833-848
[13] Frueh BE, Brown SI Transplantation of congenitally opaque corneas Br J Ophthalmol 1997 ; 81 : 1064-1069
[14] Patel HY, Ormonde S, Brookes NH, et al. The indications and outcome of paediatric corneal transplantation in New Zealand : 1991-2003 Br J Ophthalmol 2005 ; 89 : 404-408
[15] Michaeli A, Markovich A, Rootman DS Corneal transplants for the treatment of congenital corneal opacities J Pediatr Ophthalmol Strabismus 2005 ; 42 : 34-44
[16] Najjar DM, Christiansen SP, Bothun ED, Summers CG Strabismus and amblyopia in bilateral Peters anomaly J AAPOS 2006 ; 10 : 193-197
[17] Zaidman GW, Flanagan JK, Furey CC Long-term visual prognosis in children after corneal transplant surgery for Peters anomaly type I Am J Ophthalmol 2007 ; 144 : 104-108
[18] Ashar JN, Ramappa M, Vaddavalli PK Paired-eye comparison of Descemet’s stripping endothelial keratoplasty and penetrating keratoplasty in children with congenital hereditary endothelial dystrophy Br J Ophthalmol 2013 ; 97 : 1247-1249
[19] Ashar JN, Madhavi Latha K, Vaddavalli PK Descemet’s stripping endothelial keratoplasty (DSEK) for children with congenital hereditary endothelial dystrophy : surgical challenges and 1-year outcomes Graefes Arch Clin Exp Ophthalmol 2012 ; 250 : 1341-1345
[20] Comer RM, Daya SM, O’Keefe M Penetrating keratoplasty in infants J AAPOS 2001 ; 5 : 285-290
[21] Basdekidou C, Dureau P, Edelson C, et al. Should unilateral congenital corneal opacities in Peters’ anomaly be grafted? Eur J Ophthalmol 2011 ; 21 : 695-699
[22] Kim YW, Choi HJ, Kim MK, et al. Clinical outcome of penetrating keratoplasty in patients 5 years or younger : Peters anomaly versus sclerocornea Cornea 2013 ; 32 : 1432-1436
[23] Kosker M, Suri K, Rapuano CJ, et al. Long-term results of the Boston keratoprosthesis for unilateral corneal disease Cornea 2015 ; 34 : 1057-1062
[24] Bothun ED, Decanini A, Summers CG, et al. Outcome of penetrating keratoplasty for mucopolysaccharidoses Arch Ophthalmol 2011 ; 129 : 138-144
E. Bui Quoc
Le kyste dermoïde du limbe est une opacité congénitale de la cornée, périphérique, ne constituant pas classiquement une dysgénésie du segment antérieur, même si la génétique récente a montré la possible implication du gène PITX2 dans cette pathologie [1, 2]. Il s’agit d’un gène responsable de nombreuses dysgénésies du segment antérieur (voir GÉnÉTIQUE DES DYSGÉNÉSIES DU SEGMENT ANTERIEUR DE L‘ŒIL).
Le diagnostic est le plus souvent évident dès les premiers jours ou les premières semaines de vie : l’opacité de couleur blanc jaunâtre, périphérique, arrondie, surélevée et le plus souvent située au niveau du limbe temporal inférieur, étant visible à l’œil nu (Fig. 11-15).
Le kyste dermoïde du limbe est le plus souvent unilatéral et parfois bilatéral. Il a un retentissement fonctionnel (astigmatisme sévère) et esthétique. Il peut être isolé ou présent dans le cadre d’une association pathologique, comme une trisomie 18 ou un syndrome de Goldenhar ou d’autres pathologies (voir Fig. 11-16 et Tableau 11-1).
Le traitement dépend du retentissement fonctionnel et esthétique. La problématique dans le cas d’un dermoïde du limbe, même simple, est : ne pas attendre. Certes le traitement chirurgical n’est pas urgent, mais la première problématique est réfractive. Le diagnostic d’un astigmatisme fort cornéen, comme en témoigne la kératométrie, doit être établi le plus rapidement possible, afin de proposer une correction optique et d’éviter une amblyopie méridienne [3].
Après cette éventuelle correction optique, le traitement chirurgical peut être proposé, plutôt à partir de la fin de la première année de vie (l’anesthésie générale étant plus « facile » après 1 an). Diverses variantes techniques sont proposées [4, 5, 6]. Le principe est :
une exérèse de la lésion opaque qu’on clive au couteau de Crescent de la cornée sous-jacente transparente, en se méfiant d’un amincissement excessif nécessitant de réaliser, par exemple, une greffe de membrane amniotique;
et dans tous les cas une plastie conjonctivale (Fig. 11-17).
Fig. 11-15 Dermoïde du limbe.
Fig. 11-16 Dermoïdes du limbe et dermolipomes bilatéraux, anomalies des oreilles, dans le cadre d’un syndrome de Goldenhar.
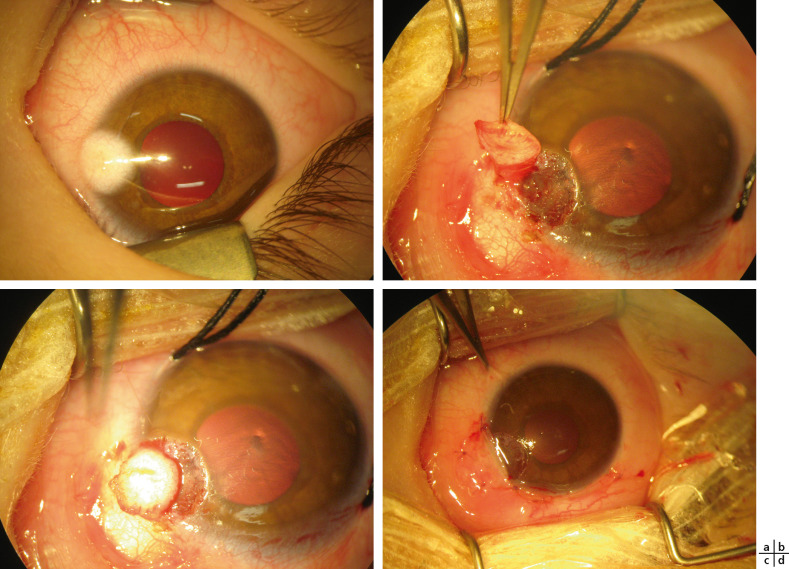
Fig. 11-17 Traitement chirurgical du dermoïde du limbe.
a. Dermoïde du limbe. b, c. Exérèse. d. Aspect final.
[1] Xia K, Wu L, Liu X, et al. Mutation in PITX2 is associated with ring dermoid of the cornea J Med Genet 2004 ; 41 : e129
[2] Doerdelmann T, Kojetin DJ, Baird-Titus JM, et al. Structural and biophysical insights into the ligand-free Pitx2 homeodomain and a ring dermoid of the cornea inducing homeodomain mutant Biochemistry 2012 ; 51 : 665-676
[3] Robb RM Astigmatic refractive errors associated with limbal dermoids J Pediatr Ophthalmol Strabismus 1996 ; 33 : 241-243
[4] Jeong J, Song YJ, Jung SI, Kwon JW New surgical approach for limbal dermoids in children: simple excision, corneal tattooing, and sutureless limboconjunctival autograft Cornea 2015 ; 34 : 720-723
[5] Lang SJ, Böhringer D, Reinhard T Surgical management of corneal limbal dermoids : retrospective study of different techniques and use of Mitomycin C Eye (Lond) 2014 ; 28 : 857-862
[6] Pirouzian A, Holz H, Merrill K, et al. Surgical management of pediatric limbal dermoids with sutureless amniotic membrane transplantation and augmentation J Pediatr Ophthalmol Strabismus 2012 ; 49 : 114-119