Pathologie inflammatoire et infectieuse : uvéites, sclérites
COORDONNÉ PAR B. BODAGHI
A-L. Remond, P. LeHoang, B. Bodaghi
L'uvéite pédiatrique est une maladie complexe ayant une grande variété d'étiologies et de présentations. Pour cette raison, elle reste un défi pour tout ophtalmologiste, de la difficulté du diagnostic à celle du traitement et de la surveillance. Les uvéites pédiatriques représentent 5 à 10 % de toutes les uvéites [1]. Les variations dans la présentation clinique, les difficultés pour réaliser un examen ophtalmologique complet, le retard diagnostique, le retentissement important de l'inflammation sur la qualité de vie, les options de gestion limitées et le fort risque d'amblyopie sont les principaux défis dans la prise en charge de cette pathologie. Elle peut conduire à une morbidité oculaire importante, avec une perte de vision majeure dans 25 à 33 % des cas.
Environ 4,3 à 6,9 sur 100 000 enfants par an développent une uvéite en Amérique du Nord. Pour les uvéites non infectieuses plus précisément, l'incidence est de 4,9 à 6,9 et la prévalence de 13 à 30 pour 100 000 enfants par an, aux États-Unis. Le début de cette atteinte est souvent insidieux. Les symptômes, lorsqu'ils sont présents, comprennent une perte de vision, une hyperhémie, une leucocorie ou encore un strabisme. L'uvéite est difficile à contrôler, en particulier pour ses formes intermédiaires, postérieures et pour les panuvéites : celles-ci répondent rarement à une thérapie topique seule et ont un fort potentiel de menace visuelle. Les formes antérieures répondent mieux au traitement local, mais celui-ci est parfois retardé en raison de l'absence de symptômes marqués et/ou gênant pour les enfants, chez lesquels la majorité des étiologies entraîne une uvéite torpide sans douleur contrairement aux formes antérieures de l'adulte. La connaissance des facteurs de risque et la surveillance systématique des sujets à risque sont deux éléments essentiels pour améliorer la prise en charge de cette pathologie.
Le traitement actuel de l'uvéite pédiatrique comprend les corticostéroïdes (par voie locale, locorégionale ou systémique), les immunomodulateurs ou immunosuppresseurs et, en présence d'un germe, les agents anti-infectieux (antibiotiques, antiviraux, etc.).
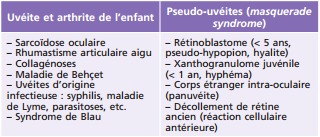
Bien que souvent idiopathique (60 % des cas), l'uvéite pédiatrique peut être associée à des étiologies auto-immunes très spécifiques de l'enfant, comme l'arthrite juvénile idiopathique (AJI), les spondylarthropathies juvéniles, ou à des maladies inflammatoires présentes aussi bien chez l'adulte comme la sarcoïdose, la maladie de Behçet, le syndrome de Vogt-Koyanagi-Harada et l'ophtalmie sympathique.
Les enfants sont particulièrement à risque de complications secondaires liées à une uvéite antérieure, comme les synéchies antérieures et postérieures, la cataracte, l'hypertonie oculaire et la neuropathie glaucomateuse, la kératopathie en bandelettes, l'œdème maculaire cystoïde, l'œdème papillaire et d'autres complications secondaires à une intervention chirurgicale. La vigilance doit être particulièrement renforcée chez le petit enfant en raison du risque très élevé et irréversible d'amblyopie.
Les uvéites antérieures représentent près de la moitié des uvéites pédiatriques (42 % ). Les principales étiologies rapportées sont l'AJI, les spondylarthropathies juvéniles, la sarcoïdose, les uvéites herpétiques, le Vogt-Koyanagi-Harada, l'ophtalmie sympathique.
L'arthrite juvénile idiopathique (AJI), anciennement appelée arthrite chronique juvénile, est la maladie rhumatismale la plus fréquente de l'enfance, avec l'uvéite comme manifestation extra-articulaire la plus commune. L'AJI est définie comme un groupe d'arthrites idiopathiques, avec une atteinte d'au moins trois articulations, survenant avant l'âge de 16 ans (pic entre 6 mois et 4 ans) et persistant pendant au moins 6 semaines. Elle touche environ 70 000 enfants aux États-Unis. Il s'agit d'une pathologie multifactorielle, avec une prédisposition génétique, idée renforcée par la présence de plusieurs membres atteints dans la même famille, et une composante environnementale, avec l'hypothèse d'un possible détonateur infectieux. La nature de l'arthrite au cours des 6 premiers mois permet de définir différents sous-groupes, et ce même si le nombre d'articulations atteintes varie par la suite (tableau 14-1 ) :
- – atteinte systémique (ou maladie de Still);
- – forme polyarticualaire (fig. 14-1 ) : ≥ 5 articulations;
- – forme oligoarticulaire : ≥ 3 et < 5 articulations.
C'est la cause la plus fréquente d'uvéite chez l'enfant et également une cause majeure de déficience visuelle acquise dans l'enfance. La prévalence de l'uvéite chez les patients atteints d'AJI varie de 4 à 38 % .
L'étiologie de la maladie est de nature auto-immune, avec la participation prédominante des cellules T CD4+ . Cependant, les méca nismes pathogènes sous-jacents demeurent flous, en particulier en ce qui concerne l'interaction entre les facteurs génétiques et environnementaux.

AAN : anticorps antinucléaire ; AJI : arthrite juvénile idiopathique ; F : femme ; FR : facteur rhumatoïde ; H : homme ; HSMG : hépatosplénomégalie ; SPA : spondylarthrite ankylosante ; UA : uvéite antérieure ; UAA : uvéite antérieure aiguë.
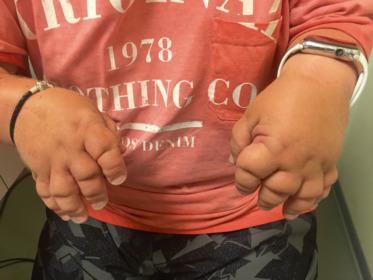
Fig. 14-1 Atteintes bilatérales sévères des mains au cours d’une forme polyarticulaire d’arthrite juvénile idiopathique.
La plupart des gènes associés à cette maladie se situent dans la région de l'antigène leucocytaire humain (human leucocyte antigen [HLA]), et cette association renforce la théorie de la maladie auto-immune. Certaines études cherchent une association entre le risque de développer une uvéite et un sous-type HLA. Chez les patients avec une forme oligoarticulaire, une uvéite antérieure chronique a été associée aux allèles HLA-DR5 et HLA-DRB1*1104. La combinaison des allèles HLA-DRB1*1104 et HLA-DPB1*0201 entraîne un risque 7,7 fois plus élevé d'uvéite chronique. HLA-B27 confère un risque accru d'uvéite antérieure aiguë. HLA-DR1 est le seul allèle HLA ayant démontré une protection contre l'uvéite antérieure chronique associée à l'AJI. Il existe néanmoins un caractère temporel à ces associations. Un allèle peut ainsi tour à tour permettre une protection ou une sensibilisation à l'AJI selon l'âge.
Au niveau cellulaire, les lymphocytes T et B sont impliqués et génèrent une réponse immunitaire dirigée contre des antigènes intra-oculaires tels que l'arrestine S, également connue sous le nom d'antigène rétinal S, le retinol-binding protein 3 et des protéines apparentées à la tyrosinase. Les biopsies oculaires montrent une prédominance de lymphocytes CD4+ plutôt que de CD8+, ainsi que des niveaux variables de lymphocytes B CD20+ [2]. Ces lymphocytes T CD4+ comprennent des cellules Th1 pro-inflammatoires (avec production d'interféron gamma) et des cellules Th17 (production d'interleukine 17), régulées par les lymphocytes T régulateurs (Treg) CD4+, CD25+, FoxP3+ naturels et induits. Il y aurait donc probablement une perte de l'homéostasie entre les différentes lignées cellulaires conduisant à une intolérance à certains « auto-antigènes » .
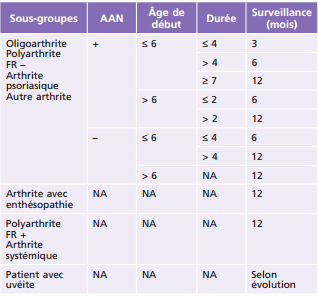
L'AJI est plus fréquente chez la fille (ratio garçon/fille = 2/3). Les filles ont également 5 fois plus de risque d'avoir une évolution plus longue. Les risques de développer des manifestations oculaires sont relativement plus fréquents chez les filles. Les principaux facteurs de risque pour le développement d'une uvéite chez les patients atteints d'AJI sont le sexe féminin, la présence d'anticorps antinucléaires (AAN, ou antinuclear antibody [ANA]), la forme oligoarticulaire, l'absence de facteur rhumatoïde et l'apparition précoce (moins de 6 ans) d'une arthrite (tableau 14-2). Ces facteurs permettent de définir un rythme de surveillance adapté (tableau 14-3).

AAN : anticorps antinucléaires ; OCT : optical coherence tomography (tomographie par cohérence optique).
AAN : anticorps antinucléaire ; FR : facteur rhumatoïde ; NA : non applicable.
La manifestation extra-articulaire la plus fréquente de l'AJI est l'inflammation intra-oculaire. Cette inflammation est typiquement une uvéite antérieure non granulomateuse, bilatérale chez 71 % des enfants, synéchiante (fig. 14-2 et fig. 14-3), d'évolution chronique. Des précipités granulomateux en graisse de mouton peuvent parfois être observés, sans remettre en question le diagnostic, en particulier chez les patients mélanodermes [3]. L'inflammation de la chambre antérieure est variable (Tyndall de 1 à 4 croix), pouvant aller parfois jusqu'à l'hypopion. On peut également observer des cellules inflammatoires dans le vitré antérieur. Les synéchies postérieures sont fréquentes. Les atteintes du segment postérieur dans l'AJI sont généralement rares mais possibles, en particulier en cas d'évolution prolongée ou de traitement insuffisant. Les principales complications responsables de baisse d'acuité visuelle sont la cataracte (due à l'inflammation et aux traitements), la kératopathie en bandelette (fig. 14-4), l'hypertonie oculaire et le glaucome secondaire, l'œdème maculaire (fig. 14-5), la membrane épirétinienne, le trou maculaire, l'hypotonie avec arrêt de sécrétion et/ou atrophie du corps ciliaire.
Cette uvéite est généralement asymptomatique et donc son dépistage chez les patients à risque présentant une AJI est primordial.
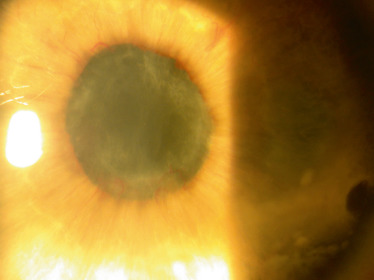
Fig. 14-2 Secclusion pupillaire au cours d’une uvéite sévère non granulomateuse associée à une arthrite juvénile idiopathique.
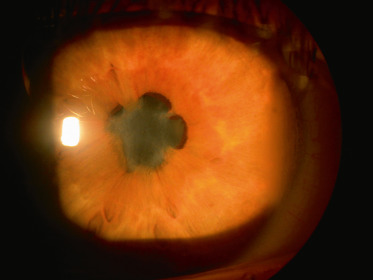
Fig. 14-3 Synéchies postérieures étendues au cours d’une arthrite juvénile idiopathique.
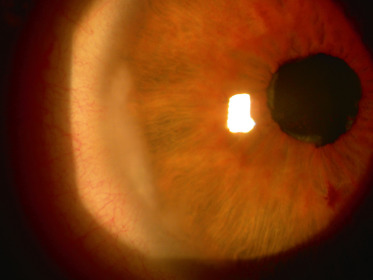
Fig. 14-4 Kératopathie en bandelettes périphérique et uvéite antérieure rhumatismale chez l’enfant.
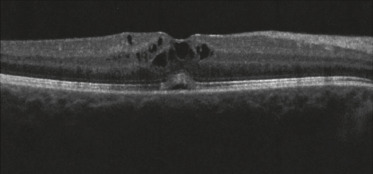
Fig. 14-5 OEdème maculaire associé à une uvéite antérieure sévère dans le cadre d’une arthrite juvénile idiopathique.
Le diagnostic de l'AJI est surtout clinique. C'est un diagnostic d'exclusion, nécessitant une histoire clinique détaillée, un examen général attentif des patients souvent en association avec un rhumatologue. Avant le début du traitement, il est important d'écarter toute cause infectieuse d'uvéite. Il n'y a aucun test biologique ultraspécifique de l'AJI. Les patients peuvent avoir une augmentation de la vitesse de sédimentation (VS), de la C-reactive protein (CRP), des leucocytes et de la numération plaquettaire. Le facteur rhumatoïde n'est pas un marqueur important pour le diagnostic de l'AJI. Il existe une forte association entre la présence d'AAN et la survenue d'une uvéite en cas d'AJI. Cependant, ce ne sont pas des marqueurs spécifiques de l'AJI, car ils peuvent être présents dans d'autres formes d'uvéite.
L'AJI est un diagnostic d'exclusion. Il est impératif de toujours penser aux pseudo-uvéites ou masquerade syndrome, étant donné la mise en jeu du pronostic vital pour certaines étiologies comme le rétinoblastome. Les principaux diagnostics différentiels sont les autres pathologies articulaires associées à une uvéite et les pseudo-uvéites (tableau 14-4).
Le traitement des uvéites comprend toujours deux volets :
- – le traitement étiologique, celui de la « cause » pour en limiter l'évolution et les récidives;
- – le traitement symptomatique, pour lutter contre l'inflammation et en limiter les conséquences, toujours désastreuses sur la fonction visuelle.
Comme pour toutes les uvéites non infectieuses, ces deux volets sont assurés dans l'AJI par les traitements anti-inflammatoires de différentes intensités. La stratégie thérapeutique en inflammation est une approche dite par paliers, c'est-à-dire avec plusieurs lignes thérapeutiques successives : la stepladder approach..
Lors d'une atteinte antérieure, le traitement de première ligne est le traitement local par collyres corticoïdes, associés ou non à des antibiotiques, en l'absence de contre-indication (CI) de la molécule associée, selon l'âge de l'enfant. Diverses molécules sont disponibles en France :
- – dexaméthasone seule : Dexafree®, Maxidex® 0,1 % ;
- – dexaméthasone en association :
- - avec tobramycine (CI < 1 an) = Tobradex® ;
- - avec néomycine (CI < 2 ans) = Chibro-cadron® ;
- - avec néomycine et la polymyxine B (CI < 2 ans) = Maxidrol ®.
Attention la tobramycine et la néomycine sont des aminosides, elles sont donc contre-indiquées chez les enfants jeunes en cours de croissance, avant 1 an et 2 ans respectivement.
Le nombre de gouttes à délivrer par jour initialement dépend de l'intensité de l'inflammation. La décroissance doit ensuite être progressive et adaptée à l'évolution.
La couverture nocturne peut être assurée par une pommade corticoïdes et antibiotiques, en respectant les précautions et les contre-indications des antibiotiques associés :
- – dexaméthasone + néomycine + polymyxine B = Maxidrol® (aminoside);
- – dexaméthasone + oxytétracycline = Sterdex® (tétracycline);
- – dexaméthasone + framycétine = Frakidex® (aminoside).
Il est nécessaire d'ajouter à ce traitement anti-inflammatoire un traitement pour prévenir la formation de synéchies. Il existe pour cela différents collyres mydriatiques, qui peuvent également avoir un effet cycloplégique antalgique supplémentaire :
- – tropicamide = Mydriaticum® 0,5 % ;
- – atropine 0,3 % , 0,5 % ou 1 % selon l'âge (0,3 % avant 3 ans, 0,5 % entre 3 et 12 ans, 1 % après 12 ans). Il faut être très vigilant à l'âge de l'enfant et à la durée du traitement lors de la prescription d'atropine, pour éviter le développement d'une amblyopie. Un traitement au long cours chez un petit enfant nécessite la prescription associée d'une addition permettant la vision de près;
- – phényléphrine (Néosynéphrine® à 2,5 % avant l'âge de 12 ans, 5 % entre 12 et 18 ans et 10 % après 18 ans) : voir chapitre 3.3.
Les injections sous-conjonctivales ou latérobulbaires de dexaméthasone sont impossibles à réaliser chez le jeune enfant sans sédation, surtout de manière répétée, mais peuvent permettre de passer un cap en renforçant le traitement local. La sédation peut être légère par recours à l'association de protoxyde d'azote et d'oxygène (Kalinox®) voire nécessiter, le plus souvent, une anesthésie générale.
Les dispositifs implantables intravitréens (corticoïdes à libération prolongée), comme l'implant de fluocinolone acétonide (Retisert®) ou l'implant de dexaméthasone (Ozurdex®), pourraient être des options chirurgicales dans certains cas réfractaires à plusieurs lignes thérapeutiques mais plutôt dans les uvéites intermédiaires et postérieures et les données manquent chez l'enfant. Retisert® libère 0,59 mg d'acétonide de fluocinolone sur plus de 30 mois; Ozurdex® libère 0,7 mg de dexaméthasone sur plus de 6 mois.
Il existe après injection de corticoïdes un risque important de développer une cataracte et un glaucome iatrogènes. Les dispositifs implantables ne sont d'ailleurs pas utilisés en France chez les enfants. Après injection sous-conjonctivale ou latérobulbaire, la surveillance tensionnelle est indispensable.
Lorsque le traitement local bien conduit ne permet pas de calmer l'inflammation, il est nécessaire d'ajouter un traitement corticoïdes par voie générale (voie orale et/ou intraveineuse) initialement. Celui-ci doit être de courte durée (≤ 3 mois à forte dose) étant donné les nombreux effets secondaires encore plus délétères chez l'enfant (retard de croissance, fonction surrénalienne, fermeture prématurée des cartilages de croissance, infection, hyperglycémie). Ce traitement est toujours prescrit en décroissance progressive. Les molécules disponibles en France sont :
- – méthylprednisolone (Solumédrol®) par voie intraveineuse : 15-30 mg/kg/j;
- – prednisolone (Solupred) ou prednisone (Cortancyl) > 6 ans (per os) :
- – dose d’attaque : 0,5-2 mg/kg/j ;
- – dose d’entretien : 0,25-0,5 mg/kg/j.
La corticothérapie per os requiert une décroissance de 10 % tous les 8 à 15 jours, avec également un possible schéma d'administration intermittente pour limiter le retard de croissance, par exemple traitement pris un jour sur deux avec double dose.
Les anti-inflammatoires non stéroïdiens (AINS) sont parfois utilisés en deuxième ligne dans les spondylarthropathies mais sont insuffisants dans l'AJI.
Lorsque les corticoïdes ne permettent pas de calmer l'inflammation ou encore que leur décroissance entraîne une récidive, l'initiation précoce d'une thérapie immunomodulatrice et d'épargne cortisonique est la clé.
Le méthotrexate, par voie orale ou en injection sous-cutanée, est généralement le premier choix dans l'AJI. Son innocuité et son efficacité sont désormais bien établies chez l'enfant. Il ne semble pas augmenter le risque de cancer. Il s'agit d'un antimétabolite, qui inhibe la synthèse de l'ADN et donc de la prolifération cellulaire. Ce traitement possède l'autorisation de mise sur le marché (AMM) dans l'AJI. Il faut généralement commencer avec une dose de 0,15 mg/kg par voie orale, 1 fois par semaine. La dose est ensuite augmentée toutes les 6 à 8 semaines jusqu'à obtenir la quiescence sans corticoïde. Les doses nécessaires sont souvent plus élevées qu'en cas d'atteinte articulaire seule. La posologie du méthotrexate étant fondée sur le poids, la dose doit donc être soigneusement modifiée selon la croissance de l'enfant. Lorsque l'inflammation persiste après plusieurs paliers, il est nécessaire de passer à la forme sous-cutanée. Si la dose est supérieure à 17,5 mg, il faut également passer à la forme sous-cutanée. Les effets secondaires possibles du méthotrexate sont la myélosuppresssion, l'hépatotoxicité et la pneumopathie interstitielle. Le méthotrexate est un analogue de l'acide folique et nécessite une supplémentation en acide folique ou folinique de 1 mg chaque jour de la semaine sauf le jour où le médicament est pris ou 5 mg, 1 fois/semaine.
Ce traitement permet une amélioration de l'uvéite dans 73 % des cas [4].
Lorsque le méthotrexate n'est pas assez efficace, d'autres immunomodulateurs peuvent être utilisés : azathioprine (Imurel®), mycophénolate mofétil (Cellcept®). La ciclosporine par voie systémique (Néoral®) n'est actuellement plus utilisée étant donné les nombreux effets secondaires. L'azathioprine (Imurel®) est modérément efficace chez les enfants et les adultes [5], mais elle est moins couramment prescrite chez les enfants, en raison des effets secondaires gastro-intestinaux plus fréquents qu'avec les autres antimétabolites. Le mycophénolate mofétil (Cellcept®) peut être efficace pour calmer l'inflammation dans plus de la moitié des cas d'uvéites réfractaires au méthotrexate, mais moins dans le cas de l'AJI.
L'infliximab (Remicade®) et l'adalimumab (Humira®), inhibiteurs du TNF-α, sont les plus efficaces de cette classe thérapeutique pour contrôler l'inflammation oculaire, et ils peuvent être utilisés seuls ou en combinaison avec une thérapie immunomodulatrice classique lorsque l'un ou l'autre est insuffisant. L'American Uveitis Society a publié des recommandations sur l'utilisation de l'infliximab et de l'adalimumab comme immunomodulateurs de deuxième ligne dans le traitement des uvéites associées à l'AJI après le méthotrexate. L'infliximab, un anticorps chimérique, est administré par voie intraveineuse, à des doses de 5 à 20 mg/kg toutes les 4 semaines, après une période d'induction. Le méthotrexate à faible dose est généralement prescrit en association avec les anti-TNF-α pour éviter l'apparition d'anticorps antichimériques. L'adalimumab, un anticorps monoclonal entièrement humanisé, peut être administré par voie sous-cutanée, à une dose de 20 ou 40 mg tous les 7 à 14 jours. L'infliximab a un effet plus rapide; l'adalimumab a moins de risque d'infection et une plus grande facilité d'administration. Selon certaines études, l'adalimumab serait peut-être légèrement plus efficace que l'infliximab [6, 7], tandis que d'autres n'indiquent pas de différence entre les deux molécules. Certains essais en cours évaluent l'efficacité de l'association méthotrexate et adalimumab contre adalimumab en monothérapie [8]. Une étude multicentrique prospective contre placebo réalisée en France et intitulée Adjuvite vient de démontrer l'efficacité de l'adalimumab et sa bonne tolérance. Ces résultats sont concordants avec ceux obtenus par une équipe anglaise au cours d'une étude similaire appelée Sycamore.
Golimumab (Simponi®) et certolizumab pegol (Cimzia®), autres anti-TNF-α, ont été utilisés pour traiter d'autres maladies auto-immunes comme la polyarthrite rhumatoïde. En ce qui concerne leur efficacité dans les uvéites, les données sont limitées à quelques cas chez l'adulte.
L'étanercept (Enbrel®), bien qu'efficace dans le traitement des manifestations rhumatologiques systémiques de l'AJI, ne doit pas être utilisé, car il est moins efficace et serait parfois responsable d'uvéite iatrogène.
Certains agents biologiques ciblant d'autres cellules immunitaires peuvent aider dans les cas d'uvéites réfractaires aux anti-TNF, mais les données actuelles, en particulier chez les enfants, sont limitées. Le tocilizumab (Roactemra®), inhibiteur de l'interleukine 6 pro-inflammatoire (IL-6), a montré une efficacité dans le traitement de ces uvéites. Le rituximab (Mabthera®), un anticorps monoclonal anti-CD20 autorisé dans le traitement de la polyarthrite rhumatoïde, des lymphomes et des leucémies, a montré une amélioration chez 7 patients sur 10 souffrant d'AJI, non contrôlée par des anticorps anti-TNF ou des agents immunomodulateurs [6]. De nombreux essais cliniques sont en cours pour évaluer l'efficacité d'autres molécules comme le sarilumab anti-IL-6, l'anakinra (Kineret®) anti-IL-1, l'ustekinumab (Stelara®) anti-IL-23 et anti-IL-12, le canakinumab (Ilaris®) anti-IL-1β et le gevokizumab anti-IL-1β.
Les agents alkylants comme le chlorambucil (Chloramonophène®) et le cyclophosphamide (Endoxan®) sont presque toujours efficaces pour réduire l'inflammation, mais ne sont plus utilisés en raison des effets secondaires potentiellement graves à long terme (tumeur maligne, dysfonction gonadique significative ou infertilité).
Le rythme de surveillance est établi selon l'atteinte ophtalmologique ou les facteurs de risque de développer une uvéite (tableau 14-3). La surveillance requiert un examen ophtalmologique complet avec la mesure de l'acuité visuelle, la prise du tonus intra-oculaire, la recherche d'une inflammation dans tous les segments de l'œil avec examen du pôle postérieur après dilatation maximale. Cet examen est associé à des examens complémentaires non invasifs répétés, tels que la tomographie en cohérence optique (optical coherence tomography [OCT]), à la recherche d'un œdème maculaire ou d'une membrane épirétinienne, et le laser flare meter seul examen permettant de mesurer objectivement de l'inflammation. Il n'est pas possible de raisonner uniquement sur une acuité visuelle ou un Tyndall, au risque de passer à côté d'un début de complications.
L'uvéite antérieure liée à l'antigène B27 est la première cause des uvéites non infectieuses chez l'adulte, et vient immédiatement derrière l'AJI chez l'enfant. L'uvéite B27 peut rester une maladie oculaire isolée ou être associée à une maladie systémique, principalement représentée par le groupe des spondylarthropathies.
Les spondylarthropathies ou spondylarthrites (SpA) sont des rhumatismes inflammatoires partageant des caractéristiques communes comme des facteurs génétiques et une atteinte privilégiée des enthèses1. L'atteinte la plus fréquente est dite axiale (atteinte du rachis, des sacro-iliaques, de la paroi thoracique antérieure) mais il existe aussi des formes périphériques, à type d'arthrite, d'oligoarthrite ou de polyarthrite (à différencier de la polyarthrite rhumatoïde). Ce groupe de pathologies comprend la spondylarthrite ankylosante (SPA), les rhumatismes associés aux maladies inflammatoires chroniques de l'intestin (MICI; rectocolite hémorragique [RCH] et maladie de Crohn), le rhumatisme psoriasique (RP), les arthrites réactionnelles, les spondylarthropathies juvéniles.
1. Enthèse : « endroit où les formations collagéniques (tendons, ligaments ou aponévroses musculaires), rentrent dans l’os. C’est une zone de transition qui passe du muscle au tendon puis au cartilage et enfin dans l’os lui-même » (https:// fr.wikipedia.org/wiki/Enth%C3%A8se).
La physiopathologie des affections liées à l'HLA-B27 est multifactorielle, faisant intervenir des facteurs génétiques, des facteurs environnementaux et des modifications de la réponse immunitaire plus ou moins liées à l'antigène.
L'antigène HLA-B27 fait partie du système HLA de classe 1, divisé en trois classes destinées à présenter des peptides antigéniques aux lymphocytes T CD8+ . Deux théories complémentaires expliqueraient le développement des atteintes rhumatismales et oculaires : celle du peptide « uvéitogène » ou « arthrogène » où la molécule HLA-B27 présenterait un ou plusieurs peptides provenant d'antigènes exprimés uniquement dans les tissus articulaires et oculaires et celle du mimétisme moléculaire avec une réaction croisée entre un antigène bactérien et des peptides du soi.
Le principal facteur de risque est la présence de l'antigène HLA-B27. Les patients atteints de SPA et porteurs d'HLA-B27 développent une uvéite dans 90 % des cas. Parmi les patients ayant eu un épisode d'uvéite liée à HLA-B27, une SpA est identifiée dans 75 % des cas.
L'uvéite est une complication fréquente dans les spondylarthropathies. C'est en général une uvéite antérieure non granulomateuse, synéchiante, plutôt unilatérale, hypotonisante (par diminution de la production d'humeur aqueuse par inflammation du corps ciliaire), récurrente, d'apparition brutale et d'évolution aiguë. Mais ses caractéristiques diffèrent légèrement selon la forme rhumatismale associée. Dans l'uvéite liée à HLA-B27, avec ou sans SPA, l'atteinte est typiquement brutale, d'évolution aiguë, antérieure et unilatérale. Elle est plus fréquente chez l'homme. L'œil est rouge, douloureux, avec un cercle périkératique. La réaction fibrineuse (Tyndall protéique) prédomine sur la réaction cellulaire (Tyndall cellulaire), définissant une uvéite dite « plastique » . La présence d'un hypopion n'est pas exceptionnelle. Dans l'uvéite associée à un RP ou une MICI, l'uvéite peut être antérieure et intermédiaire, bilatérale, chronique, touchant le plus souvent des femmes. La fréquence de l'uvéite est également beaucoup plus importante en association avec la SPA qu'avec les autres formes.
Le diagnostic ophtalmologique est clinique, conforté par la présence de l'antigène HLA-B27 et/ou d'une atteinte générale connue. Il comprend essentiellement les caractéristiques de l'uvéite et la recherche de signes extra-ophtalmologiques en faveur d'une SPA, d'un RP, d'une MICI ou d'une arthrite réactionnelle : signes digestifs, cutanés et articulaires. Il est plus aisé lorsque l'atteinte générale est déjà connue. Il ne faut cependant pas méconnaître une infection, surtout en présence d'une atteinte récurrente, unilatérale et toujours du même côté.
L'approche se fait également par paliers avec :
- – le traitement local et locorégional de la(des) poussée(s);
- – le traitement systémique par corticoïdes, en cas d'insuffisance du traitement local et/ou de récurrences trop fréquentes;
- – le traitement systémique par AINS. Peu efficaces dans l'AJI, ces molécules le sont pour prévenir les récurrences dans les spondylarthropathies. Couramment utilisées chez l'adulte, elles nécessitent certaines précautions chez l'enfant, et certaines molécules sont contre-indiquées en dessous de 15 ans. Les anti-inflammatoires peuvent être associés aux corticoïdes ou aux immunomodulateurs au besoin [9]. Ils sont néanmoins moins utilisés depuis l'avènement de nombreux agents biologiques;
- – le traitement systémique par immunomodulateurs. La démarche thérapeutique et les immunomodulateurs utilisés dans les spondylarthropathies sont les mêmes que dans l'AJI.
La surveillance est clinique et paraclinique et adaptée à l'évolution. Les examens non invasifs sont à privilégier, d'autant plus chez l'enfant. La prise en charge est multidisciplinaire, entre les ophtalmologistes, les rhumatologues et les pédiatres.
La sarcoïdose, ou maladie de Besnier-Boeck-Schaumann (communément dénommée BBS) ou lymphogranulomatose bénigne, est une maladie inflammatoire systémique de cause inconnue, avec une atteinte préférentielle des poumons mais qui peut atteindre tous les organes, y compris la peau et les ganglions lymphatiques. La sarcoïdose se manifeste par la présence de granulomes (amas de cellules inflammatoires). Les conséquences sont variables selon les organes touchés.
Le diagnostic de certitude est histologique. La biopsie d'un granulome est donc nécessaire sauf s'il existe un syndrome de Löfgren suffisant pour poser le diagnostic (atteinte articulaire et arthralgie, adénopathies médiastinales, fièvre, érythème noueux, anergie tuberculinique). Il faut prélever le granulome le plus accessible (peau, glande salivaire, adénopathie, bronche) et l'examen anatomopathologique met en évidence un granulome épithélioïde et gigantocellulaire sans nécrose caséeuse.
Les sarcoïdoses d'apparition précoce sont des formes génétiques comme le syndrome de Blau ou le syndrome CINCA (chronic infantile neurological cutaneous and articular = pathologie chronique infantile avec manifestations neurologique, cutanée et articulaire).
Le syndrome de Blau, ou sarcoïdose d'apparition précoce (early onset sarcoïdosis [EOS]), est une maladie auto-inflammatoire monogénique rare granulomateuse, causée par une mutation autosomique dominante dans le gène NOD2/CARD15 Ce syndrome se réfère aux formes familiales et sporadiques de la maladie auto-inflammatoire granulomateuse pédiatrique, tandis que d'autres utilisent les termes de syndrome de Blau, sporadique ou héréditaire. Cette atteinte se traduit cliniquement chez le nourrisson et le petit enfant par une éruption granulomatose de la peau (dermatite granulomateuse : érythème maculopapulaire du tronc et des extrémités), une polyarthrite (synovite et ténosynovite des articulations périphériques) et une uvéite récurrente. Cette uvéite est antérieure, postérieure ou totale. L'âge médian de survenue est de 4,4 ans dans 80 % des cas. Non traitée, la maladie peut causer une cécité et un handicap moteur sévère du fait de l'arthrite exubérante avec des déformations articulaires.
Tous les types d'uvéites peuvent se voir, d'une simple uvéite antérieure unilatérale à une panuvéite bilatérale sévère. L'uvéite antérieure est uni- ou bilatérale, typiquement chronique, mais aussi aiguë, granulomateuse, avec des précipités en graisse de mouton, parfois de très grande taille et répartis souvent sur la moitié inférieure, des nodules iriens (de Busacca et de Koeppe). Le Tyndall de chambre antérieure est variable, les synéchies iridocristalliniennes et l'hypertonie oculaire fréquentes. La trabéculite est une forme particulière d'uvéite antérieure, évocatrice de la sarcoïdose. Il s'agit d'une infiltration de l'angle iridocornéen par des nodules sarcoïdosiques. L'inflammation peut être minime et l'hypertonie très importante. L'évolution se fait vers l'apparition de goniosynéchies et vers un glaucome secondaire.
L'examen du segment postérieur est essentiel et doit être minutieux à la recherche d'une uvéite intermédiaire et/ou postérieure associées posant souvent l'indication d'un traitement par voie générale. Il est parfois nécessaire de réaliser une angiographie à la recherche de vascularites périphériques et de conséquences ischémiques.
La démarche thérapeutique est identique à celle proposée dans les spondylarthropathies (voir plus haut).
Le syndrome TINU (tubulo-interstitial nephritis and uveitis) a été décrit pour la première fois par Dobrin et al. en 1975. C'est une maladie rare, majoritairement idiopathique, et sous-diagnostiquée en cas d'uvéite chez l'enfant. Il s'agit d'un diagnostic d'élimination. L'âge médian d'apparition est de 15 ans avec une prépondérance féminine (ratio garçon/fille = 1/3). La cause de l'évolution de la maladie à médiation immunitaire reste largement incertaine. Les symptômes courants sont un malaise, une hyperthermie, une anorexie et une perte de poids. L'uvéite est le plus souvent antérieure (80 % des uvéites), non granulomateuse, le plus souvent bilatérale (d'emblée dans plus de 50 % des cas, ou dans environ 70 % des cas dans un délai de 20 mois) et récurrente. Elle peut être accompagnée d'une atteinte intermédiaire et postérieure avec une hyalite, une papillite, un œdème maculaire cystoïde, une choriorétinite et une choroïdite multifocale. Un délai médian de 1 mois est observé entre le début des signes généraux et les signes ophtalmologiques mais certaines observations rapportent des signes ophtalmologiques jusqu'à 2 mois avant et 14 mois après les manifestations systémiques.
Il existe un syndrome inflammatoire avec une VS supérieure à 50 mm (première heure) dans 90 % des cas, une possible anémie inflammatoire, une hypergammaglobulinémie; ces signes sont régressifs en cas guérison. L'hyperéosinophilie sanguine est inconstante (un tiers des cas environ), les complexes immuns circulants, le facteur rhumatoïde et/ou les facteurs antinucléaires sont exceptionnellement retrouvés. Une anergie tuberculinique est fréquente.
Les cas suspects de TINU doivent être adressés aux néphrologues. Les analyses urinaires montrent une glycosurie, une protéinurie, une cétonurie et une hématurie microscopique. La nécrose de l'épithélium rénal du tubule, l'œdème interstitiel et l'infiltrat lymphocytaire peuvent être mis en évidence à la biopsie rénale. La néphrite se résout habituellement spontanément, bien que les corticoïdes par voie orale soient souvent indiqués pour prévenir les conséquences. La néphropathie chronique survient dans 11 % des cas. Selon la gravité et le site de l'inflammation, le traitement de l'uvéite sera local, locorégional ou général.
Il n'y a pas de consensus thérapeutique clair. Cependant la mise en route rapide d'une corticothérapie à fortes doses a été suivie d'une évolution favorable dans la grande majorité des cas publiés.
Le syndrome TINU est un diagnostic d'élimination. Il faut rechercher d'autres pathologies pouvant entraîner une atteinte rénale et oculaire : infections (syphilis, toxoplasmose, tuberculose, histoplasmose, brucellose, mononucléose infectieuse) ou maladies systémiques (sarcoïdose, syndrome de Sjögren, lupus, maladie de Wegener). La sarcoïdose et le syndrome de Sjögren en particulier sont les plus susceptibles de confusion avec un syndrome TINU.
L'évolution habituelle de la néphrite se fait vers la guérison sous traitement corticoïde mais des guérisons spontanées ont été rapportées, ainsi que des aggravations sous corticoïdes. Chez l'adulte, l'insuffisance rénale persiste ou s'aggrave parfois quand le diagnostic a été tardif, par développement d'une fibrose interstitielle. Divers immunosuppresseurs (azathioprine, ciclosporine, méthotrexate, mycophénolate mofétil) ont également été utilisés avec succès chez quelques patients avec uvéite réfractaire à la corticothérapie.
La maladie de Vogt-Koyanagi-Harada (VKH), initialement décrite comme une uvéo-méningo-encéphalite, est une maladie auto-immune systémique touchant les tissus riches en mélanocytes, tels que l'uvée, l'oreille interne, les méninges, la peau et les cheveux. Elle survient en général vers 30-40 ans. Le VKH chez l'enfant est rare et principalement confiné à quelques cas rapportés dans la littérature.
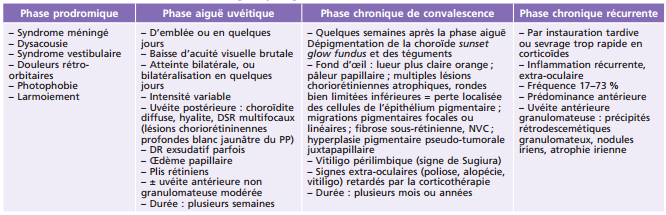
Cette atteinte évolue généralement en quatre phases :
- – une phase prodromique avec les symptômes neurologiques;
- – une phase aiguë uvéitique;
- – une phase chronique de convalescence;
- – une phase chronique avec récurrences.
Ces phases sont plus ou moins marquées et pas toujours toutes présentes, compliquant le diagnostic. Il est donc possible de voir un patient avec une uvéite antérieure aiguë, uni- ou bilatérale, non granulomateuse au cours de la phase aiguë uvéitique, ou granulomateuse au cours de la phase chronique récurrente. Il faut alors rechercher les autres symptômes généraux et ophtalmologiques. Le diagnostic est clinique et rassemble un certain nombre d'éléments (tableau 14-5). Le nombre de critères présents permet de poser un diagnostic de certitude (forme complète avec cinq critères, fortement probable avec quatre, ou probable avec trois).
Le traitement de la poussée d'uvéite est local ou locorégional. Le traitement par voie générale doit être mis en place rapidement, surtout en présence d'atteinte postérieure mais aussi antérieure sévère pour limiter les récurrences et stopper l'évolution vers la phase de convalescence. Ce traitement repose sur l'utilisation des corticoïdes à fortes doses par voie générale dans un premier temps, puis d'immunomodulateurs permettant une épargne cortisonique, d'autant plus importante que le patient est jeune. Les traitements de premier choix sont le mycophénolate mofétil, possible à partir de 2 ans, et le méthotrexate en raison du recul très important concernant cette molécule. On peut également avoir recours aux anti-TNF-α et aux anti-interleukines en présence de formes réfractaires, d'évolution défavorable avec les traitements précédents.
DR : décollement rétinien ; DSR : décollement séreux rétinien ; NVC : néovaisseaux choroïdiens ; PP : pôle postérieur.
Les uvéites antérieures virales sont essentiellement dues aux virus du groupe herpès : le virus herpès simplex 1 et 2 (herpes simplex virus [HSV-1 et 2]), le virus zona-varicelle (VZV), le cytomégalovirus (CMV) et le virus Epstein-Barr (Epstein-Barr virus [EBV]). Elles représentent environ 7,69 % des uvéites pédiatriques [10]. Il existe deux tableaux cliniques différents.
Les virus HSV, VZV sont très répandus chez l’adulte, jusqu’à 80 % pour HSV et 90 % pour VZV. Ces virus sont transmis par contact direct. L’infection primaire a lieu au niveau de la peau et des muqueuses, et la latence dans les ganglions trigéminés et lombosacrés. La varicelle est la manifestation clinique de la primo-infection à VZV, et le zona, celle de sa réactivation. Les uvéites surviennent rarement au cours de la primo-infection, mais plutôt lors de la réactivation.
L’uvéite peut être associée à une kératite (épithéliale, stromale ou endothéliale), la précédant ou survenant au décours. Une uvéite antérieure survient dans 10 % des kératites herpétiques, en particulier la kératite disciforme.
L’atteinte est classiquement unilatérale. Les précipités rétrocornéens peuvent être fins ou granulomateux en graisse de mouton, de localisation inférieure ou diffuse (fig. 14-6). L’inflammation peut être très importante allant parfois jusqu’à l’hypopion et/ou une hypertonie oculaire. L’examen clinique révèle souvent une hypoesthésie cornéenne, plus profonde en cas de VZV. D’autres signes peuvent être observés tels qu’une épisclérite ou une sclérite. Une atrophie irienne est le témoin d’une poussée ancienne. Celleci est plutôt sectorielle avec HSV et plus diffuse avec VZV.
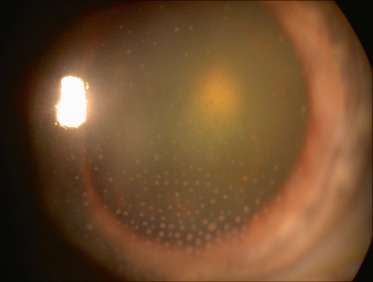
Fig. 14-6 Uvéite antérieure granulomateuse unilatérale liée à une atteinte herpétique.
Le diagnostic est clinique. Toute uvéite hypertensive unilatérale doit faire penser à une atteinte herpétique. On recherche des lésions associées suggestives d’une atteinte herpétique telles que des lésions cutanées, une kératite, une hypoesthésie cornéenne, une atrophie irienne. L’examen du segment postérieur doit être minutieux, à la recherche de nécrose rétinienne. Lorsque la kératite est absente, le diagnostic est plus difficile.
La sérologie est rarement informative. Seule la négativité présente un intérêt, celui d’exclure le diagnostic, sauf chez les patients immunodéprimés.
Le seul diagnostic de certitude, lorsque celui-ci est nécessaire, se fait par la mise en évidence du virus, par polymerase chain reaction (PCR), ou de la synthèse intra-oculaire d’anticorps, sur un prélèvement d’humeur aqueuse (ponction de chambre antérieure [PCA]). La sensibilité de cet examen est variable et beaucoup plus faible en présence d’une atteinte seulement antérieure qu’en cas de rétinite nécrosante. Le résultat négatif n’exclut donc pas le diagnostic.
Le traitement repose sur un traitement étiologique antiviral et un traitement anti-inflammatoire.
Ce traitement vise à contrôler la réplication virale mais également à lutter contre les phénomènes inflammatoires et vasculaires qui lui sont associés. Tous les antiviraux existant actuellement sont virostatiques et non virucides. Il est nécessaire d’instaurer donc un traitement d’attaque suivi par une phase d’entretien à adapter selon l’évolution clinique et paraclinique.
Bien que l’aciclovir (Zovirax®) ait été le traitement de référence, les trois dernières décennies ont vu l’introduction de nombreux traitements antiviraux, aussi bien par voie orale, tels que le valaciclovir (Zelitrex®), le famciclovir (Oravir®) et le valganciclovir (Rovalcyte®) plus spécifique du CMV. Les deux molécules utilisées en première intention dans l’uvéite antérieure sont l’aciclovir et le valaciclovir. L’aciclovir est efficace sur l’HSV-1 et 2 et le VZV. L’aciclovir est un analogue de la guanine, il doit être phosphorylé à trois reprises (triphosphorylé) pour être actif en se liant à l’acide désoxyribonucléique (ADN)-polymérase virale. La spécificité de l’action de l’aciclovir vient donc du fait qu’il est phosphorylé par la thymidine kinase du virus de l’herpès et non par la thymidine kinase de la cellule de l’hôte, si bien que l’on trouve beaucoup d’aciclovir sous forme active, c’est-à-dire triphosphatée, dans les cellules infectées et pas dans les cellules non infectées. Au plan pharmacocinétique, la biodisponibilité de l’aciclovir par voie orale est faible. Le valaciclovir est la prodrogue de l’aciclovir, car il est constitué d’une molécule d’aciclovir estérifiée par la L-valine, ce qui lui confère une biodisponibilité par voie orale nettement supérieure à celle de l’aciclovir. L’administration orale de valaciclovir permet chez l’adulte, lorsqu’on utilise la dose maximale de 3 g/j, d’être aussi efficace qu’une administration par voie veineuse d’aciclovir.
Les doses d’aciclovir et de valaciclovir sont à adapter au poids et la fonction rénale de l’enfant : dose d’aciclovir 10 mg/kg toutes les 8 heures ; forme disponible en solution buvable ou par voie intraveineuse chez l’enfant.
Le traitement repose sur l’utilisation de corticoïdes. Ce traitement est administré par voie locale et/ou locorégionale selon l’intensité de la réaction inflammatoire en chambre antérieure. Il doit être associé à un traitement de prévention des synéchies, et ce par des collyres mydriatiques. Il ne doit pas être administré immédiatement en présence d’une kératite associée et ne doit jamais être poursuivi sans un traitement antiviral de couverture.
Le CMV peut être responsable d’uvéites antérieures. L’uvéite antérieure à CMV peut se présenter sous plusieurs formes cliniques différentes : l’uvéite dite « à CMV » , le syndrome de Posner-Schlossman et parfois une hétérochromie irienne de Fuchs. L’uvéite dite à CMV possède les mêmes caractéristiques que celle à HSV et à VZV en dehors des synéchies postérieures.
Le syndrome de Posner-Schlossman est une cyclite hypertonisante récurrente décrite par Posner et Schlossman en 1948. Le CMV a ensuite été mis en évidence dans l’humeur aqueuse au cours de ce syndrome. Ce syndrome apparaît plutôt chez l’adulte de 20 à 50 ans et est rare chez l’enfant. Il s’agit d’une uvéite antérieure aiguë, non granulomateuse avec parfois quelques précipités granulomateux « sentinelles » , unilatérale, hypertonisante et récurrente. L’inflammation est souvent modérée, le tonus très élevéet la douleur quasi absente. La réponse est rapidement favorable aux collyres corticoïdes et hypotonisants. Le pronostic à long terme est réservé en cas de récidives. Le principal diagnostic différentiel est le glaucome par fermeture de l’angle.
Les synéchies antérieures et postérieures, la cataracte, l'hypertonie oculaire et la neuropathie glaucomateuse, la kératopathie en bandelette, l'œdème maculaire cystoïde, l'œdème papillaire, les complications secondaires à une intervention chirurgicale et l'amblyopie sont les principales complications liées à une uvéite antérieure chez l'enfant.
La chirurgie de la cataracte chez l'enfant atteint d'uvéite antérieure a été longtemps controversée. L'implantation en chambre postérieure proposée à la fin du siècle dernier par BenEzra [11] n'a été validée que récemment. En effet, la garantie du succès postopératoire dépend du contrôle de l'inflammation oculaire (fig. 14-7 à 14-9); l'absence d'un traitement médical agressif et l'implantation chez un enfant âgé de moins de 6 ans et atteint d'AJI étaient synonymes autrefois de catastrophe allant jusqu'à l'explantation. Désormais, les critères favorables à l'implantation sont de mieux en mieux connus et la chirurgie est donc possible [12,13]. Néanmoins, grâce au diagnostic plus précoce de l'atteinte oculaire, avant la survenue de complications significatives, et à l'avènement des anti-TNF-α, l'incidence de la cataracte a significativement diminué et l'âge des enfants nécessitant une chirurgie a augmenté. Il est donc possible d'espérer que la cataracte ne soit plus une complication significative de ce type d'uvéite d'ici 5 à 10 ans.
Le glaucome reste redoutable lorsqu'il s'autonomise et échappe au traitement médical [14]. Les corticoïdes jouent un rôle néfaste largement démontré et doivent être prescrits de façon très prudente, à la dose la plus faible possible. L'évaluation de l'inflammation au tyndallomètre laser permet d'éviter tout surdosage délétère à plus ou moins long terme. En cas d'échec du traitement antihypertenseur, la chirurgie reste indiquée en préférant la trabéculectomie étant donné le risque de synéchies antérieures ou postérieures au cours de la maladie. Malheureusement, les valves de drainage deviennent nécessaires dans les formes réfractaires.
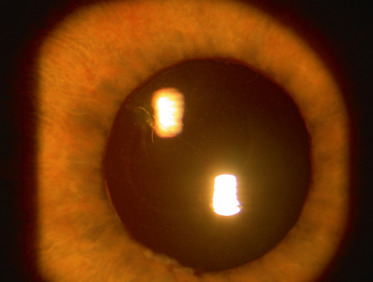
Fig. 14-7 Aspect postopératoire après phacoémulsification avec rhexis postérieur et implantation dans le sac au cours d’une uvéite de type arthrite juvénile idiopathique avec un excellent résultat postopératoire grâce à un traitement médical bien conduit.

Fig. 14-8 Prolifération de cellules géantes au niveau de l’implant intra-oculaire survenue après l’arrêt brutal du traitement anti-inflammatoire.
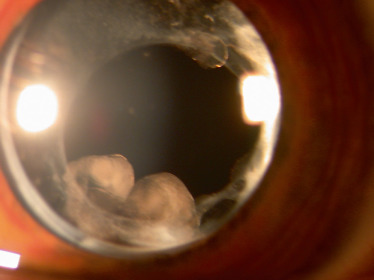
Fig. 14-9 Perles d’Elschnig apparues plusieurs années après une phacoexérèse chez une fille atteinte d’arthrite juvénile idiopathique.
L'uvéite pédiatrique diffère dans certains aspects de celle de l'adulte. L'association avec une maladie systémique et la présentation de celle-ci diffère de manière significative. L'uvéite chez l'enfant est souvent asymptomatique, malgré la gravité de l'inflammation et de la diminution de la vision. Ainsi, l'uvéite est souvent découverte de manière fortuite, et les complications irréversibles sont déjà présentes. Les traitements systémiques tels que les corticostéroïdes, les plus couramment utilisés dans le traitement de l'inflammation intra-oculaire, ont des effets secondaires délétères sur l'os en développement et donc sur la croissance, et doivent être utilisés judicieusement. Les immunosuppresseurs doivent être utilisés avec une surveillance encore plus étroite des patients. Les résultats des interventions chirurgicales dans la gestion des diverses complications de l'uvéite pédiatrique, comme la cataracte, le décollement de rétine, le glaucome, sont souvent insuffisants. La prévention reste essentielle, permettant une identification et un traitement rapide de toute complication menaçante.
[1] Angeles-Han ST, Rabinovich CE Uveitis in children Curr Opin Rheumatol ( 2016 ) :28: 544-549
[2] Clarke SLN, Sen ES, Ramanan AV Juvenile idiopathic arthritis-associated uveitis Pediatr Rheumatol Online J ( 2016 ) :14: 27 p
[3] Keenan JD, Tessler HH, Goldstein DA Granulomatous inflammation in juvenile idiopathic arthritis-associated uveitis J AAPOS ( 2008 ) :12: 546-550
[4] Sen ES, Dick AD, Ramanan AV Uveitis associated with juvenile idiopathic arthritis Nat Rev Rheumatol ( 2015 ) :11: 338-348
[5] Pasadhika S, Kempen JH, Newcomb CW Azathioprine for ocular inflammatory diseases Am J Ophthalmol ( 2009 ) :148: 500-509 e2
[6] Heiligenhaus A, Minden K, Föll D, Pleyer U Uveitis in juvenile idiopathic arthritis Dtsch Ärztebl Int ( 2015 ) :112: 92-100 i
[7] Simonini G, Taddio A, Cattalini M Prevention of flare recurrences in childhood-refractory chronic uveitis: an open-label comparative study of adalimumab versus infliximab Arthritis Care Res ( 2011 ) :63: 612-618
[8] Ramanan AV, Dick AD, Benton D A randomised controlled trial of the clinical effectiveness, safety and cost-effectiveness of adalimumab in combination with methotrexate for the treatment of juvenile idiopathic arthritis associated uveitis (SYCAMORE Trial) Trials ( 2014 ) :15: 14 p
[9] Naz S, Mushtaq A, Rehman S Juvenile rheumatoid arthritis J Coll Physicians Surg Pak ( 2013 ) :23: 409-412
[10] Engelhard SB, Bajwa A, Reddy AK Causes of uveitis in children without juvenile idiopathic arthritis Clin Ophthalmol Auckl NZ ( 2015 ) :9: 1121-1128
[11] BenEzra D Cataract surgery and intraocular lens implantation in children Am J Ophthalmol ( 1996 ) :121: 224-226
[12] Terrada C, Julian K, Cassoux N Cataract surgery with primary intraocular lens implantation in children with uveitis: long-term outcomes J Cataract Refract Surg ( 2011 ) :37: 1977-1983
[13] Guindolet D, Dureau P, Terrada C Cataract surgery with primary lens implantation in children with chronic uveitis Ocul Immunol Inflamm ( 2016 ) :6: 1-7
[14] Wang Q, Wang J, Fortin E, Hamel P Trabeculectomy in the treatment of pediatric uveitic glaucoma J Glaucoma ( 2016 ) :25: 744-749
C. Couret
Selon la standardization of uveitis nomenclature (SUN), le terme d'uvéite intermédiaire (UI) caractérise toute inflammation du vitré antérieur, du corps ciliaire et de la rétine périphérique, associée ou non à une cause infectieuse ou une pathologie inflammatoire systémique; la pars planite constitue une entité d'UI spécifique, définie par l'association d'œufs de fourmis, de banquises et l'absence d'étiologie [1]. Néanmoins, certains auteurs soulignent l'ambiguïté de cette définition, mêlant diagnostic topographique et étiologique [2], Il semblerait plus clair de définir l'UI comme une inflammation de la pars plana, de la rétine et de la choroïde périphérique sans lésion focale rétinienne ou choroïdienne en arrière de la base du vitré, et de réserver le terme de pars planite à une entité topographique pure, caractérisée par la présence de banquises, sans préjuger de l'origine inflammatoire, infectieuse ou idiopathique de l'atteinte.
L'incidence des UI varie de 1,2 à 4 cas pour 100 000 habitants en France et aux États-Unis [3,4] et les UI représentent 5 à 26,7 % des uvéites de l'enfant [3,5,6]. Elles touchent principalement les enfants entre 6 et 10 ans [7,8] et sont le plus souvent bilatérales, même si souvent asymétriques, avec parfois seulement quelques cellules dans le vitré de l'œil le moins atteint, La majorité des UI de l'enfant demeure idiopathique [9,10] et d'étiopathogénie inconnue.
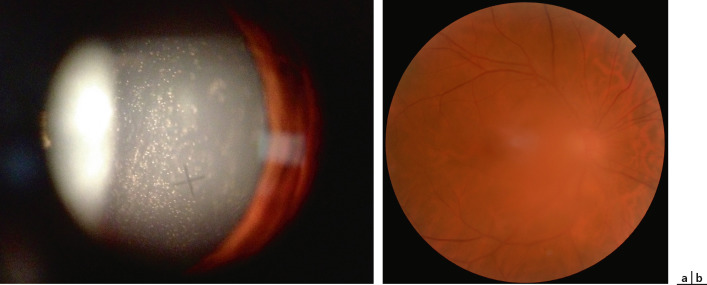
Fig. 14-10 Hyalite au cours d’une uvéite intermédiaire idiopathique.
a. Tyndall cellulaire vitréen antérieur. b. Haze vitréen.
Myodésopsies et flou visuel sont les signes d'appel les plus fréquemment retrouvés mais l'UI peut être asymptomatique et de découverte fortuite. Il existe parfois une baisse d'acuité visuelle importante secondaire à une hyalite dense (fig. 14-10) ou un œdème maculaire.
Le diagnostic d'UI repose sur l'examen attentif du vitré qui révèle la présence de cellules vitréennes, mieux vues et quantifiables dans le vitré antérieur, d'un haze vitréen dont l'intensité est déterminée par la visibilité du fond d'œil (tableau 14-6) [1], d'œufs de fourmis prenant la forme d'agrégats blanc-jaune dans le vitré moyen et en périphérie inférieure (fig. 14-11), et d'exsudats de la pars plana, appelés banquises (fig. 14-12). L'examen du fond d'œil peut également retrouver des périphlébites rétiniennes périphériques, un œdème papillaire et un œdème maculaire. L'inflammation antérieure de contiguïté est souvent modeste voire absente.
En raison du caractère asymptomatique de l'inflammation à la phase initiale, a fortiori chez les jeunes enfants, les UI peuvent causer une atteinte fonctionnelle irréversible en cas de diagnostic tardif et de lésions tissulaires sévères [11]. Les complications les plus souvent rencontrées sont l'œdème maculaire cystoïde (25,7 à 47,7 % des cas selon les études [12-14] avec un délai moyen de survenue de 5,7 ans [15]) et la cataracte. Suivent les condensations vitréennes, qui, lorsqu'elles sont antérieures, peuvent être confondues avec une cataracte sous-capsulaire postérieure chez les plus jeunes enfants, la papillite, rarement à l'origine d'une atrophie optique, et les vascularites. D'autres complications cécitantes sont moins fréquentes. La kératopathie en bandelette témoigne très volontiers d'un début d'inflammation dans l'enfance même si elle peut survenir à tout âge. Le glaucome est rare. La cause la plus fréquente des hémorragies intravitréennes de l'enfant est l'UI [16]. En effet, les néovaisseaux papillaires et sous-rétiniens péripapillaires, témoins d'une inflammation sévère, ainsi que la néovascularisation des banquises, peuvent saigner. Les décollements de rétine, rhegmatogènes, tractionnels ou exsudatifs, volontiers inférieurs, sont rares. On retrouve également quelques rétinoschisis inférieurs, spécificité pédiatrique, dont la physiopathologie est discutée (gliose à l'ora serrata induite par les banquises ou angiogenèse périphérique en rapport avec l'inflammation chronique), quelques trous maculaires et ectopies maculaires. Enfin, il peut coexister un certain degré d'amblyopie si des complications obstruant l'axe visuel surviennent et persistent pendant la période de maturation corticale visuelle principalement avant 6 ans.
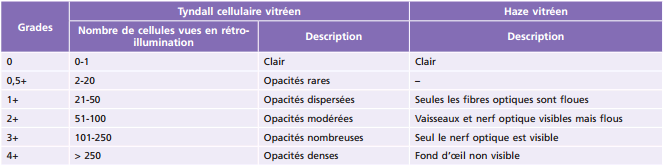
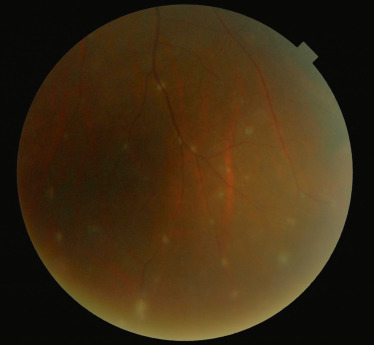
Fig. 14-11 OEufs de fourmis en inférieur au cours d’une uvéite intermédiaire idiopathique.
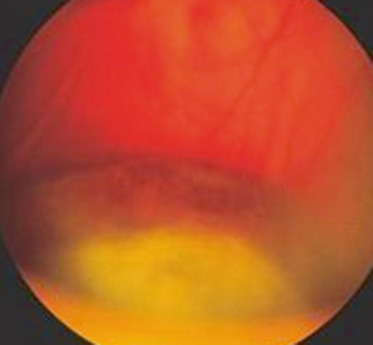
Fig. 14-12 Banquise inférieure au cours d’une uvéite intermédiaire idiopathique.
Les rétinophotographies en couleurs, réalisées à la phase initiale, sont indispensables pour documenter le haze vitréen et les complications rétiniennes associées aux UI afin de mieux apprécier l'évolution et la réponse au traitement des UI (fig. 14-13).
Examen rapide et non invasif, la tomographie par cohérence optique (optical coherence tomography [OCT]) offre de nombreux éléments diagnostiques, pronostiques et de suivi. Si elle permet de visualiser les cellules vitréennes à l'interface vitréomaculaire, elle permet surtout de réaliser une évaluation quantitative et qualitative objective des complications maculaires : œdèmes maculaires (œdème maculaire cystoïde, épaississement maculaire diffus ou périfovéolaire, décollement séreux rétinien) et leurs séquelles à type d'atrophie maculaire et syndromes de l'interface vitréomaculaire (membrane épirétinienne, syndrome de traction vitréomaculaire, trou maculaire). L'altération de la ligne IS/OS (inner segment/outer segment) serait un marqueur pronostique péjoratif de récupération visuelle [17].
L'angiographie à la fluorescéine a été remplacée par l'OCT pour le diagnostic et le suivi de l'œdème maculaire inflammatoire associé aux UI pédiatriques, mais elle garde une place de choix pour mettre en évidence et suivre la réponse thérapeutique des lésions vasculaires et papillaires inflammatoires fréquemment associées aux UI pédiatriques : périphlébites et capillarites volontiers périphériques et dans la région des banquises, néovaisseaux rétiniens et papillite parfois infracliniques. Quant à l'angiographie au vert d'indocyanine, elle ne trouve sa place que pour éliminer une inflammation choroïdienne infraclinique à la phase initiale.
L'échographie peut s'avérer utile lorsqu'une membrane cyclitique, une cataracte, une hyalite dense ou une hémorragie intravitréenne rendent l'examen du fond d'œil impossible ou incomplet. Plus performante que l'échographie conventionnelle (8 à 10 Hz), l'échographie ultrasound biomicroscopy (UBM) permet de visualiser avec précision la région de la pars plana et de mettre en évidence banquises, membranes cyclitiques, membranes vitréennes et tractions vitréorétiniennes périphériques.
Fig. 14-13 Uvéite intermédiaire idiopathique bilatérale chez une enfant de 12 ans avec baisse d’acuité visuelle et myodesopsies droites.
Acuité visuelle : 4/10 oeil droit (OD) et 10/10 oeil gauche (OG) = hyalite et oedème maculaire cystoïde droits, oedème papillaire droit et capillarite diffuse bilatérale. a.OCT OD : hyalite et oedème maculaire cystoïde. b. OCT OG normal. c. Angiofluorographie OD pôle postérieur : oedème papillaire droit. d. Angiographie OD périphérie temporale : capillarite diffuse droite. e. Angiofluorographie OG pôle postérieur : normal. f. Angiographie OG périphérie temporale : capillarite diffuse gauche.
Le diagnostic des UI est clinique mais l'absence d'œuf de fourmi ou de banquise peut faire errer le diagnostic, surtout si le fond d'œil périphérique, notamment avec indentation sclérale, n'est pas bien visualisé.
L'UI chronique ne doit pas être confondue avec l'AJI, d'évolution également chronique et asymptomatique et partageant les mêmes complications. L'atteinte antérieure au premier plan et la présence d'une arthrite chronique peuvent redresser le diagnostic. De même, il faut savoir différencier l'UI de l'iridocyclite de Fuchs, unilatérale, et caractérisée par une hyalite à gros grains associée à des précipités rétrocornéens diffus parfois stellaires, une atrophie irienne avec ou sans hétérochromie et l'absence d'œdème maculaire. Enfin, une pathologie systémique, inflammatoire, infectieuse ou tumorale doit être éliminée (tableau 14-7).
L'évolution naturelle des UI est variable : 10 % d'évolution spontanément favorable, 59 % d'évolution chronique avec exacerbations et 31 % d'évolution chronique avec peu d'exacerbations, et 70 % de complications cécitantes. Le pronostic dépend avant tout de la sévérité et de la durée de l'inflammation vitréenne. Paroli et al. ont identifié les facteurs de mauvais pronostic suivants : l'âge de début inférieur à 10 ans, le sexe masculin, la durée d'évolution au moment du diagnostic supérieure à 3 ans, la présence de cellules en chambre antérieure, la sévérité du haze vitréen, la présence d'œufs de fourmi, de banquises et/ou d'un œdème maculaire [12]. Seuls des contrôles réguliers permettent d'établir une stratégie thérapeutique adaptée.
Le traitement des UI n'est pas consensuel. Actuellement, l'indication repose plus sur la sévérité de l'UI que sur l'acuité visuelle elle-même [11]. En effet, il semblerait qu'un traitement précoce et agressif offre une meilleure récupération visuelle qu'un traitement initié tardivement lorsque l'acuité visuelle atteint le pallier de 5/10 avec des lésions tissulaires souvent déjà irréversibles. La mise en route d'un traitement est indiquée lorsqu'il existe un œdème maculaire cystoïde, une hyalite altérant la fonction visuelle (baisse d'acuité visuelle et/ou altérations campimétriques), une vascularite, des banquises importantes ou des lésions témoignant d'une inflammation chronique destructrice telles qu'une kératopathie en bandelettes ou une cataracte.
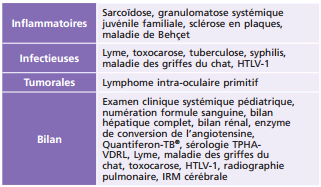
HTLV-1 : human T cell leukemia virus 1 ; IRM imagerie par résonance magnétique ; TPHA-VDRL : treponema pallidum haemagglutinations assay-venereal disease research laboratory.
Lorsqu'il est indiqué, le traitement de référence des UI reste la corticothérapie. Les corticoïdes topiques ne sont efficaces que sur l'atteinte antérieure, a fortiori chez les patients phaques. Les corticoïdes péri-oculaires (injections sous-conjonctivales de 4 à 12 mg de dexaméthasone, voire sous-ténoniennes de 40 mg de triamcinolone) ou intra-oculaires (implants de 700 µg dexaméthasone) sont efficaces [18] et utilisables lorsque l'atteinte est très asymétrique ou pour passer un cap inflammatoire avec certaines limites à connaître : efficacité temporaire pour une pathologie chronique évoluant sur plusieurs années; injections à réaliser sous anesthésie générale chez le jeune enfant; cataracte et glaucome cortisonique préjudiciables à terme. En cas d'atteinte sévère, a fortiori si elle est bilatérale, il est préférable d'initier le traitement par une corticothérapie systémique (prednisone 1 à 1,5 mg/kg/j) diminuée progressivement voire d'y associer 3 injections intraveineuses de méthylprednisolone (10 mg/kg/j – max. 1 g/j – sur 3 jours ou sur 6 jours à raison d'un jour sur deux) initialement si l'on souhaite une action anti-inflammatoire rapide et intense.
En deuxième intention en cas de cortico-dépendance à un seuil inacceptable, il convient d'associer un traitement immunosuppresseur d'épargne cortisonique : méthotrexate, azathioprine ou mycophénolate mofétil, selon les habitudes des cliniciens ophtalmologistes et pédiatres. Le long délai d'efficacité de ces molécules nécessite de poursuivre la corticothérapie initialement et d'attendre au minimum 8 semaines avant de juger qu'elles sont inefficaces.
Enfin, en troisième intention, les biothérapies peuvent être utilisées : anti-TNF-α (infliximab ou adalimumab) ou interféron α ou β. L'utilisation des anti-TNF-α doit être prudente en raison du risque d'association entre UI et sclérose en plaques (SEP) et du risque accru de pathologies démyélinisantes avec les anti-TNF-α.
Dans certains cas, il peut être nécessaire de réaliser une vitrectomie lorsqu'il existe une baisse d'acuité visuelle en rapport avec des condensations vitréennes, une hémorragie intravitréenne, un décollement de rétine, une membrane épirétinienne, une traction vitréomaculaire ou un œdème maculaire résistant au traitement médical. La chirurgie de la cataracte peut être réalisée dans de bonnes conditions avec de bons résultats si les règles suivantes sont respectées : contrôle inflammatoire préopératoire de 3 mois minimum, chirurgie soigneuse, implant acrylique hydrophobe dans le sac, contrôle inflammatoire per- et postopératoire.
La cryothérapie des banquises a été supplantée par les traitements immunosuppresseurs et immunomodulateurs. Quant à la photocoagulation laser, elle garde une place dans le traitement des néovaisseaux rétiniens périphériques, en adjonction au traitement médical.
- ➤ Les UI de l’enfant, le plus souvent idiopathiques, de nature chronique et insidieuse, peuvent être cécitantes. Elles sont plus sévères que chez l’adulte et le retard de prise en charge diagnostique et thérapeutique peut être à l’origine d’atteintes anatomiques et fonctionnelles irréversibles
- ➤ Un traitement précoce et agressif des atteintes inflammatoires sévères semble plus efficace pour préserver la fonction visuelle de ces enfants atteints d’UI qu’un traitement initié lorsque l’acuité visuelle chute en dessous de 5/10.
- ➤ Une prise en charge médicale par étapes avec corticoïdes, immunosuppresseurs puis immunomodulateurs puis chirurgicale avec vitrectomie, voire photocoagulation laser semble efficace pour limiter les dégâts tissulaires et fonctionnels liés aux complications des UI.
HTLV-1 (human T-cell leukemia virus-1) est un provirus à ADN intégré au génome des lymphocytes T, responsable de leucémies et de lymphomes et impliqué dans l'œil sous une forme infiltrative ou par infections opportunistes notamment à CMV. Dans les zones endémiques (Caraïbes, Afrique centrale, Japon), il a été démontré que la séroprévalence pour HTLV-1 était plus élevée dans le groupe des uvéites idiopathiques, notamment chez l'enfant, que dans le groupe contrôle [19]. Ces résultats suggèrent l'existence d'une entité clinique distincte : l'uvéite liée à HTLV-1, avec 3 % d'enfants et d'adolescents atteints.
Classiquement responsable d'hémopathies malignes, le virus est également associé à des atteintes inflammatoires pulmonaires [20] ou thyroïdiennes comme la maladie de Basedow [21].
Les manifestations oculaires sont variées et diffèrent d'une zone d'endémie à une autre. La maladie a un début souvent brutal uni- ou bilatéral avec des myodésopsies, une gêne ou un flou visuel. Il s'agit en effet d'une uvéite intermédiaire dans 60 % des cas, suivie d'une panuvéite (23 % ), d'une uvéite antérieure (14 % ) puis plus rarement d'une vascularite rétinienne à vitré clair (2 % ) [22]. L'atteinte typique se présente comme une hyalite modérée à sévère (opacités vitréennes quasi constantes), accompagnée d'une réaction inflammatoire antérieure minime (précipités rétrocornéens en graisse de mouton dans 18 % des cas, fibrine et hypopion < 2 % des cas) et d'une vascularite rétinienne modérée non occlusive, sans lésion choriorétinienne. Il peut exister une hyperhémie papillaire d'évolution favorable avec la régression de l'inflammation et un œdème maculaire cystoïde. Alors que les vascularites sont plus fréquentes au Japon, les kératoconjonctivites sèches et kératites interstitielles atteignent plutôt les populations des Caraïbes et d'Afrique centrale. Le pronostic est globalement bon même si l'uvéite à HTLV-1 récidive dans 50 à 60 % des cas. Dix pour cent des patients présentent néanmoins à terme une acuité visuelle inférieure à 3/10 en raison de complications : cataracte (20 % ), glaucome (16 % ), membrane épirétinienne (6 % ) ou œdème maculaire cystoïde (4-6 % ).
II s'agit d'un diagnostic d'élimination chez des enfants dont la sérologie HTLV-1 est positive. Sarcoïdose, maladie de Behçet et pars planite doivent être exclues. De même, le fond d'œil attentif doit rechercher des lésions rétiniennes en faveur d'une nécrose rétinienne virale ou d'une candidose.
Les corticoïdes sont très efficaces : sous forme topique en cas d'atteinte antérieure, péri- ou intra-oculaire lorsque l'atteinte intermédiaire est unilatérale et invalidante, et per os lorsque l'atteinte est bilatérale (0,5 mg/kg/j avec diminution progressive sur 3 à 4 semaines).
- ➤ L’uvéite intermédiaire dysimmunitaire est la manifestation la plus fréquente des atteintes inflammatoires oculaires à HTLV-1. Néanmoins, il existe une variabilité phénotypique d’une zone d’endémie à l’autre avec une prédominance de vascularites au Japon.
- ➤ L’uvéite à HTLV-1 est un diagnostic d’élimination de pronostic globalement bon chez des enfants avec une sérologie positive, cortico-sensible mais récidivante.
La borréliose de Lyme est une zoonose non alimentaire fréquente présente en France2, en Europe, en Amérique du Nord et dans les régions tempérées de l'Asie. L'incidence non rare et l'existence d'un traitement préventif des complications tissulaires à la phase tardive justifient l'existence d'une surveillance épidémiologique assurée par le réseau Sentinelles et le Centre national de référence des Borrelia(centre hospitalo-universitaire de Strasbourg) [23].
En Europe, la bactérie en cause dans la maladie de Lyme est principalement Borrelia burgdorferi sensu lato (B. sl.). En France, elle est transmise d'avril à novembre par morsure de tique du genre Ixodes ricinus, vivant dans les forêts de feuillus, sous-bois, pâturages, prairies, zones boisées péri-urbaines et parcs en ville.
2. Voir http://invs.santepubliquefrance.fr/Dossiers-thematiques/Maladies-infectieuses/ Maladies-a-transmission-vectorielle/Borreliose-de-lyme/Donnees-epidemiologiques.
L'atteinte systémique est caractérisée par trois stades évolutifs de survenue très variable. L'érythème chronique migrant (ECM) du stade 1 apparaît au niveau du site d'inoculation dans 44 à 77 % des cas [23] entre le 3 et le 30 jour après la morsure de tique (passée inaperçue dans 50 % des cas) et disparaît spontanément en 3 à 4 semaines. L'atteinte neurologique, cutanée, musculosquelettique, cardiaque ou oculaire, du stade 2 survient entre quelques jours à quelques semaines après la morsure en l'absence de traitement antibiotique. Après plusieurs mois d'évolution sans traitement, 10 % des patients développent une arthrite, manifestation la plus fréquente du stade 3, parfois associée à une atteinte neurologique, cutanée ou oculaire [24].
Le spectre de l'inflammation oculaire attribuée à la maladie de Lyme est large et mal connu. L'uvéite est le plus souvent bilatérale et survient aux stades tardifs de la maladie : stades 2 (1 % d'atteinte ophtalmologique [23]) et 3.
On peut observer des conjonctivites aiguës modérées au stade 1, des kératites stromales ou interstitielles au stade 3 ou des uvéites par atteinte pathogène directe du spirochète et/ou par inflammation secondaire à la réaction immunitaire contre lui. L'uvéite antérieure peut être granulomateuse ou non, avec des précipités rétrocornéens granulomateux et/ou des nodules iriens [25]. L'uvéite intermédiaire est l'atteinte oculaire liée à la maladie de Lyme la plus fréquente. La hyalite, parfois importante, peut être accompagnée d'une réaction inflammatoire antérieure (fig. 14-14). d'une papillite et d'une vascularite. Elle répond habituellement bien aux antibiotiques. Une atteinte inflammatoire postérieure est également possible sous la forme d'une neurorétinite ou d'une choroïdite multifocale uni- ou bilatérale, avec ou sans hyalite, prenant l'aspect d'une épithéliopathie en plaques et parfois compliquée d'un décollement de rétine exsudatif. On retrouve enfin des vascularites rétiniennes artérielles ou veineuses ou capillarites parfois occlusives. Quelques cas de panuvéites et d'endophtalmies ont été décrits. Par ailleurs, la maladie de Lyme peut être à l'origine de myosites orbitaires et, plus communément, d'atteintes neuro-ophtalmologiques associant paralysie faciale périphérique, paralysie du VI (directe ou indirecte par hypertension intracrânienne) et plus rarement paralysie du III, du IV ou du V. On retrouve enfin des atteintes papillaires, papillites, névrites optiques, neuropathies optiques ou œdème papillaire de stase, et des anomalies pupillaires telles qu'une mydriase paralytique, une pupille tonique d'Addie ou un syndrome de Claude-Bernard-Horner [26].
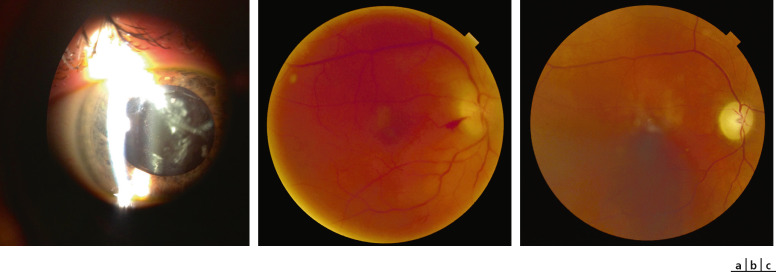
Fig. 14-14 Maladie de Lyme OD.
a. Lampe à fente initiale : Tyndall cellulaire et remaniements vitréens antérieurs. b. Rétinographie non mydriatique initiale : haze 2+ et oedème papillaire avec hémorragie péripapillaire temporale. c. RNM après traitement par ceftriaxone par voie intraveineuse (IV), méthylprednisolone IV 3 jours et relais prednisone per os : régression de la hyalite et atrophie optique séquellaire.
Le diagnostic de la maladie de Lyme repose sur un faisceau d'arguments épidémiologique et anamnestique (morsure de tique dans une région endémique et ECM, clinique, atteinte inflammatoire oculaire compatible), et biologique (sérologies positives). La sérologie ELISA (enzyme-linked immunosorbent assay) doit être confirmée par Western blot afin de limiter les faux positifs. Il est également possible de mettre en évidence B. burgdorferi par PCR [23] dans l'humeur aqueuse ou dans le vitré, même si la sensibilité n'est pas parfaite chez l'enfant, et la nécessité de pratiquer ces prélèvements sous anesthésie générale limite leur réalisation.
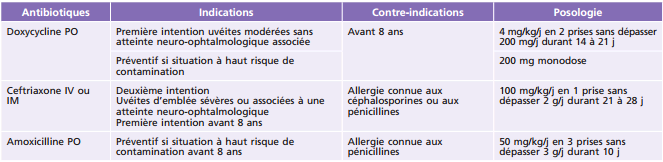
IM : (voie) intramusculaire ; IV : (voie) intraveineuse ; PO : per os.
Sarcoïdose, tuberculose et syphilis, entités uvéitiques inflammatoires également polymorphes doivent être éliminées.
L'antibiothérapie générale est adaptée à l'expression clinique systémique et oculaire de la maladie de Lyme [25]. En cas d'uvéite, il n'y a pas de schéma clairement établi. Doxycycline et ceftriaxone peuvent être utilisées (tableau 14-8). Une corticothérapie locale ou générale sera associée selon l'importance de la réaction inflammatoire oculaire [23].
En termes de prévention, il est recommandé d'éviter les zones infestées de tiques et de procéder à un examen cutané des enfants au retour de balades pour ablation précoce de la tique en cas de morsure. En cas de morsure de tique chez l'enfant, une antibioprophylaxie sera initiée au cas par cas dans les situations à haut risque de contamination (piqÛres multiples, long délai d'attachement, fort taux d'infestation connu) (tableau 14-8).
La toxocarose est une zoonose parasitaire cosmopolite transmise par Toxocara canis. La prévalence de la maladie est d'autant plus grande que les conditions socio-économiques sont défavorables [27,28]. En effet, les enfants se contaminent par géophagie ou ingestion d'aliments souillés par les déjections canines et par contact avec les chiots. Ils ingèrent des œufs embryonnés contenant des larves infestantes qui, une fois libérées dans l'intestin grêle, migrent à travers la paroi intestinale où elles gagnent la circulation lymphatique et sanguine. L'atteinte oculaire se fait probablement via la circulation choroïdienne. Une réaction inflammatoire granulomateuse à éosinophiles intense autour de la larve stoppe le cycle parasitaire mais est à l'origine de lésions tissulaires notamment tractionnelles destructrices. On peut observer un syndrome de larva migrans viscéral ou oculaire. La majorité des patients avec une sérologie positive sont asymptomatiques et ceux atteints d'une forme oculaire ne présentent que rarement des signes systémiques (fig. 14-15).
La maladie de Lyme est une zoonose fréquente à l’origine d’atteintes inflammatoires oculaires polymorphes, notamment uvéales intermédiaires chez l’enfant, de diagnostic difficile (faisceau d’arguments épidémiologique, anamnestique, clinique et biologique) mais accessibles au traitement antibiotique.
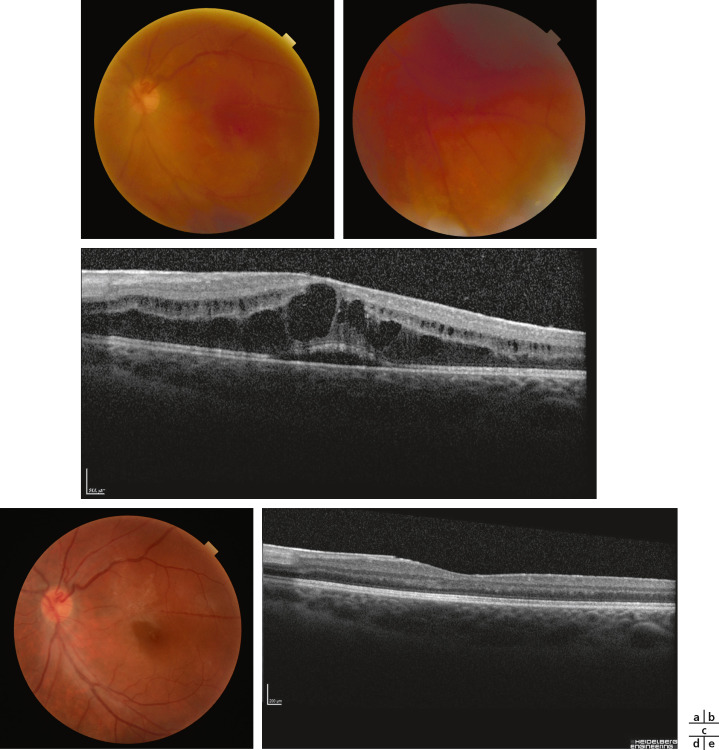
Fig. 14-15 Toxocarose OG chez un enfant de 10 ans de découverte fortuite au cours d’un examen systématique. Acuité visuelle 1,2/10 cataracte sous-capsulaire postérieure au contact d’une hyalite antérieure « tractionnelle » .
a, b. RNM initiale : haze 3+ et granulome rétinien unique avec tractions vitréorétiniennes périphériques non vues : RNM du pôle postérieur (a) ; RNM de la périphérie inférotemporale (b). c. OCT initiale : hyalite, membrane épirétinienne et oedème maculaire cystoïde. d, e. RNM (d) et OCT (e) après traitement par albendazole et corticoïdes IV, puis relais per os, puis injection sous-conjonctivale de triamcinolone = résolution de l’oedème maculaire avec membrane épirétinienne et remaniements vitréens tractionnels postérieurs minimes, tractions vitréorétiniennes périphériques sur foyer cicatriciel sans décollement de rétine non vues.
Le syndrome de larva migrans viscéral touche les enfants de moins de 3 ans, allant du syndrome pseudo-grippal isolé au tableau multiviscéral parfois mortel, accompagné d'une hyperéosinophilie.
À distance de l'atteinte systémique, la toxocarose oculaire est typiquement unilatérale chez des enfants plus âgés, jusqu'à l'adolescence [29], et en bonne santé, révélée principalement par une leucocorie, un strabisme, une baisse d'acuité visuelle ou des myodésopsies [30].
Le granulome rétinien, plus souvent périphérique que postérieur dans l'aire maculaire, rarement dans le nerf optique, prend l'aspect d'une masse blanche ou grise, souvent non hémorragique, en relief et entourée de plis rétiniens radiaires vers la papille. La hyalite est constante mais variable, caractérisée par des cordages vitréens vers le vitré adjacent, la papille, voire la macula. L'inflammation antérieure est minime et la pression intra-oculaire normale. Plus rarement et chez les plus jeunes enfants, on retrouve une hyalite dense et un décollement de rétine exsudatif, un hypopion, de la fibrine en chambre antérieure et une membrane cyclitique sur un œil habituellement blanc et non douloureux, un tableau d'endophtalmie chronique faisant suspecter un rétinoblastome ou un corps étranger intra-oculaire.
L'inflammation régresse spontanément lorsque la larve meurt mais l'évolution cicatricielle fibrotique est à l'origine de lésions tissulaires irréversibles potentiellement cécitantes : cicatrice du pôle postérieur, œdème maculaire, membrane épirétinienne, plis rétiniens, décollement de rétine, membrane néovasculaire choroïdienne, remaniements vitréens denses, cataracte, glaucome néovasculaire et phtise.
La toxocarose oculaire peut également se présenter sous d'autres formes : une neurorétinite diffuse unilatérale subaiguë (diffuse unilateral subacute neuroretinitis [DUSN]) [31] à la phase initiale de laquelle on peut identifier le nématode sous-rétinien mobile; une neuropathie optique inflammatoire; une occlusion de l'artère centrale de la rétine; une uvéite intermédiaire isolée. Les atteintes antérieures sont possibles : kératites et kératoconjonctivites, cataracte et nématode intracristallinien. Rarement, la toxocarose entraîne des hémorragies rétiniennes isolées, une iridocyclite, des granulomes en chambre antérieure ou des lésions orbitaires.
Le diagnostic est clinique, confirmé par la positivité des sérologies oculaires ELISA et Western blot dans l'humeur aqueuse ou le vitré. Le test ELISA sérique est positif à des taux très variables et peut être négatif sans exclure le diagnostic. L'hyperéosinophilie n'est parfois présente que dans les liquides oculaires. L'examen parasitologique des selles est inutile car toujours négatif.
L'échographie en mode B montre une masse solide hyperréflective parfois calcifiée [32] sans corps étranger, des tractions vitréennes tendues entre le granulome et le pôle postérieur voire un décollement de rétine. L'UBM visualise mieux l'aspect pseudokystique ou le granulome de la pars plana. L'imagerie par résonance magnétique (IRM) orbitaire retrouve un hypersignal T1 et T2. La tomodensitométrie n'apporte pas d'élément utile.
Le diagnostic différentiel principal est le rétinoblastome, qui peut ou pouvait être à l'origine de nombreuses énucléations dans la crainte de ce diagnostic en cas de formes évoluées avec décollement de rétine. En faveur du rétinoblastome, on retient : l'âge de révélation souvent plus précoce (< 2 ans); le caractère éventuellement bilatéral; l'aspect bulleux du décollement de rétine; l'extension sous-rétinienne de la lésion sans remaniement vitréen ni cataracte; les calcifications intratumorales; un hypersignal en IRM seulement en T1. La toxocarose peut également mimer une maladie de Coats, une rétinopathie du prématuré, une vitréorétinopathie exsudative familiale, une hyperplasie du vitré primitif, une toxoplasmose ou une endophtalmie endogène ou post-traumatique (corps étranger intra-oculaire).
La larve oculaire est inaccessible au traitement médical. Vivante, elle poursuit sa migration intra-oculaire puis meurt et reste en place. La corticothérapie systémique précoce, adaptée à la sévérité de l'inflammation et associée à l'albendazole (tableau 14-9) [33], diminue la hyalite et les séquelles tractionnelles. La tentative d'ablation chirurgicale de la larve est vaine car souvent difficile d'accès, génératrice d'inflammation et source de complications. Seul le décollement de rétine justifie une chirurgie endoculaire à la phase active. La vitrectomie, diagnostique et thérapeutique, concerne surtout les complications afin de libérer les tractions vitréorétiniennes pouvant être à l'origine d'un décollement de rétine ou d'une membrane épirétinienne invalidante.
Le traitement préventif de cette infection intra-oculaire cécitante repose sur les mesures d'hygiène des mains et de préparation des fruits et légumes, la limitation de la géophagie dans les régions chaudes et humides, la vermifugation des animaux domestiques et le respect des règles d'hygiène des espaces publiques de jeux (bac à sable, etc.).
- ➤ La toxocarose est une zoonose endoculaire grave à laquelle il faut penser devant une hyalite ou un granulome rétinien unilatéral de l’enfant.
- ➤ Le traitement curatif est peu efficace et la prévention via l’hygiène des mains et la vermifugation des chiots est primordiale.
- ➤ Le principal diagnostic différentiel est le rétinoblastome (fig. 14-16).
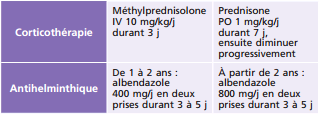
IV : (voie) intraveineuse ; PO : per os.
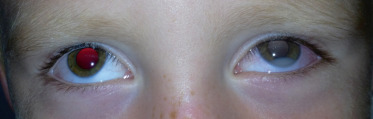
Fig. 14-16 Rétinoblastome gauche : hypopion et leucocorie gauches chez un enfant de 5 ans adressé pour uvéite.
La convexité vers le haut de l’hypopion correspond à un amas de cellules tumorales.
[1] Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature (SUN) Working Group Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop Am J Ophthalmol ( 2005 ) :140: 509-516
[2] Khairallah M Are the Standardization of the Uveitis Nomenclature (SUN) Working Group criteria for codifying the site of inflammation appropriate for all uveitis problems? Limitations of the SUN Working Group classification Ocul Immunol Inflamm ( 2010 ) :18: 2-4
[3] Gritz DC, Wong IG Incidence and prevalence of uveitis in Northern California; the Northern California Epidemiology of Uveitis Study Ophthalmology ( 2004 ) :111: 491-500 discussion 500
[4] Vadot E Epidemiology of intermediate uveitis : a prospective study in Savoy Dev Ophthalmol ( 1992 ) :23: 33-34
[5] Nikkhah H, Ramezani A, Ahmadieh H Childhood pars planitis; clinical features and outcomes J Ophthalmic Vis Res ( 2011 ) :6: 249-254
[6] Ganesh SK, Bala A, Biswas J Pattern of pediatric uveitis seen at a tertiary referral center from India Ocul Immunol Inflamm ( 2016 ) :24: 402-409
[7] Rosenberg KD, Feuer WJ, Davis JL Ocular complications of pediatric uveitis Ophthalmology ( 2004 ) :111: 2299-2306
[8] Heinz C, Schoonbrood S, Heiligenhaus A Intermediate uveitis in children and young adults: differences in clinical course, associations and visual outcome Br J Ophthalmol ( 2014 ) :98: 1107-1111
[9] Romero R, Peralta J, Sendagorta E, Abelairas J Pars planitis in children : epidemiologic, clinical, and therapeutic characteristics J Pediatr Ophthalmol Strabismus ( 2007 ) :44: 288-293
[10] Jabs DA, Nussenblatt RB, Rosenbaum JT Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop Am J Ophthalmol ( 2005 ) :140: 509-516
[11] Ozdal PC, Berker N, Tugal-Tutkun I Pars Planitis : epidemiology, clinical characteristics, management and visual prognosis J Ophthalmic Vis Res ( 2015 ) :10: 469-480
[12] Paroli MP, Spinucci G, Monte R Intermediate uveitis in a pediatric Italian population Ocul Immunol Inflamm ( 2011 ) :19: 321-326
[13] Prieto JF, Dios E, Gutierrez JM Pars planitis : epidemiology, treatment, and association with multiple sclerosis Ocul Immunol Inflamm ( 2001 ) :9: 93-102
[14] Kump LI, Cervantes-Castaneda RA, Androudi SN, Foster CS Analysis of pediatric uveitis cases at a tertiary referral center Ophthalmology ( 2005 ) :112: 1287-1292
[15] Donaldson MJ, Pulido JS, Herman DC Pars planitis : a 20-year study of incidence, clinical features, and outcomes Am J Ophthalmol ( 2007 ) :144: 812-817
[16] Lauer AK, Smith JR, Robertson JE, Rosenbaum JT Vitreous hemorrhage is a common complication of pediatric pars planitis Ophthalmology ( 2002 ) :109: 95-98
[17] Pakzad-Vaezi K, Or C, Yeh S, Forooghian F Optical coherence tomography in the diagnosis and management of uveitis Br J Ophthalmol ( 2016 nov 29 )
[18] Tomkins-Netzer O, Lightman S, Drye L Outcome of treatment of uveitis macular edema : the multicenter uveitis steroid treatment trial 2-year results Ophthalmology ( 2015 ) :122: 2351-2359
[19] Mochizuki M, Watanabe T, Yamaguchi K Uveitis associated with human T-cell lymphotropic virus type I Am J Ophthalmol ( 1992 ) :114: 123-129
[20] Sugimoto M, Mita S, Tokunaga M Pulmonary involvement in human T-cell lymphotropic virus type-I uveitis : T-lymphocytosis and high proviral DNA load in bronchoalveolar lavage fluid Eur Respir J ( 1993 ) :6: 938-943
[21] Yamaguchi K, Mochizuki M, Watanabe T Human T lymphotropic virus type 1 uveitis after graves disease Br J Ophthalmol ( 1994 ) :78: 163-166
[22] Takahashi T, Takase H, Urano T Clinical features of human T-lymphotropic virus type 1 uveitis : a long-term follow-up Ocul Immunol Inflamm ( 2000 ) :8: 235-241
[23] Société de pathologie infectieuse de langue française. Borréliose de Lyme : démarches diagnostiques, thérapeutiques et préventives. 16 Conférence de consensus en thérapeutique anti-infectieuse, décembre 2006.
[24] Donta ST Late and chronic Lyme disease Med Clin North Am ( 2002 ) :86: 341-349 vii
[25] Caspers L, Lefèbvre P, Kadz B, Willermain F Uvéites dues à la maladie de Lyme Dir. Les uvéites:Masson ( 2010 ) 139-146
[26] Wender JT, Cunningham , Emmet T Borreliosis (Lyme disease) Ocular Inflammation:Springer Verlag ( 2016 ) 1071-1079
[27] Centers for Disease Control and Prevention (CDC) Ocular toxocariasis–United States, 2009-2010 MMWR Morb Mortal Wkly Rep ( 2011 ) :60: 734-736
[28] Carden SM, Meusemann R, Walker J Toxocara canis : egg presence in Melbourne parks and disease incidence in Victoria Clin Experiment Ophthalmol ( 2003 ) :31: 143-146
[29] Woodhall D, Starr MC, Montgomery SP Ocular toxocariasis : epidemiologic, anatomic, and therapeutic variations based on a survey of ophthalmic subspecialists Ophthalmology ( 2012 ) :119: 1211-1217
[30] Oréfice F, dos Reis Veloso CE, Alves Costa R, Lambert Oréfice J Toxocariasis Ocular Inflammation:Springer Verlag ( 2016 )
[31] Sabrosa NA, de Souza EC Nematode infections of the eye: toxocariasis and diffuse unilateral subacute neuroretinitis Curr Opin Ophthalmol ( 2001 ) :12: 450-454
[32] Morais FB, Maciel AL, Arantes TE Ultrasonographic findings in ocular toxocariasis Arq Bras Oftalmol ( 2012 ) :75: 43-47
[33] Urban B, Bakunowicz-Łazarczyk A, Michał S Clinical features, the effectiveness of treatment and function of vision organ in children and adolescents with ocular toxocariasis Klin Oczna ( 2008 ) :110: 364-366
L. Kodjikian
La toxoplasmose est une maladie parasitaire caractérisée par son tropisme oculaire. Elle constitue l'étiologie la plus fréquente des uvéites postérieures dans le monde [1]. L'agent pathogène, Toxoplasma gondii, est un protozoaire intracellulaire obligatoire, probablement le parasite le plus répandu dans le monde. Ce dernier affecte les humains et les animaux, les félins en l'occurrence, le chat étant son hôte définitif. Il s'agit d'une maladie infectieuse congénitale ou acquise. La maladie peut se transmettre par ingestion, inhalation, transplantation d'organe, transfusion sanguine ou transmission transplacentaire. La toxoplasmose peut être responsable de séquelles visuelles. Une perte de la vision centrale peut apparaître en cas d'atteinte de la macula et/ou du nerf optique et serait présente chez environ un patient sur quatre [2]. Une atteinte du champ visuel existerait quant à elle dans deux tiers des cas [2].
Il faut distinguer l'infection post-natale acquise de l'infection prénatale congénitale. L'infection acquise représenterait environ deux tiers des toxoplasmoses oculaires pour un tiers d'infections congénitales. Cependant, en dehors de quelques situations particulières, il semble impossible de différencier cliniquement une toxoplasmose congénitale d'une toxoplasmose acquise, les tests sérologiques ne pouvant pas les distinguer à la phase chronique de l'infection [3].
Les principaux symptômes de la toxoplasmose oculaire sont les corps flottants, la baisse d'acuité visuelle ou le scotome central, si l'enfant est suffisamment grand pour les identifier. Une uvéite granulomateuse unilatérale, classiquement hypertensive, avec synéchies iridocristalliniennes peut être retrouvée. Au fond d'œil, l'inflammation du vitré peut être importante et dense, en masquant ainsi les détails. Le foyer de rétinite peut alors être à peine perceptible au travers la hyalite, expliquant la classique métaphore du « phare dans le brouillard » . La cicatrice rétinochoroïdienne avec des bords nets et parfois pigmentée apparaît après 6 à 8 semaines d'évolution. Dans la toxoplasmose congénitale, on peut noter un aspect parfois pseudo-colobomateux de la cicatrice (fig. 14-17). Le diagnostic de certitude ne doit pas retarder la mise en route du traitement si celui-ci est urgent avec une présomption clinique de toxoplasmose oculaire forte. La sérologie est quasiment inutile puisqu'elle ne peut ni affirmer une toxoplasmose oculaire ni l'in firmer, des faux négatifs de la sérologie ayant été rapportés [4]. L'angiographie peut apporter une aide, en montrant une hyperfluorescence centripète (fig. 14-18). L'OCT montre classiquement au stade actif une hyperréflectivité des couches internes de la rétine neurosensorielle avec en regard des cellules inflammatoires intravitréennes, représentées par des points hyperréflectifs et souvent un décollement du vitré localisé (fig. 14-19). Seule l'analyse de l'humeur aqueuse a une valeur, avec calcul du coefficient de charge immunitaire (coefficient de Witmer-Desmonts, considéré comme positif au-delà de 3) et/ou réalisation d'un Western blot(immunoblot) à la recherche d'une synthèse intra-oculaire d'anticorps spécifiques, l'amplification génique à la recherche du génome toxoplasmique par PCR n'étant pas utilisée en pédiatrie car pas suffisamment sensible chez l'enfant et l'adulte immunocompétents. Néanmoins, d'une part ces tests peuvent comporter des faux négatifs dans les 3 à 4 premières semaines suivant le début de la maladie, les anticorps spécifiques n'étant pas encore présents à un taux suffisamment détectable; d'autre part, la ponction de chambre antérieure est un geste invasif, notamment chez l'enfant de jeune âge. Le diagnostic en particulier chez l'enfant demeure clinique.
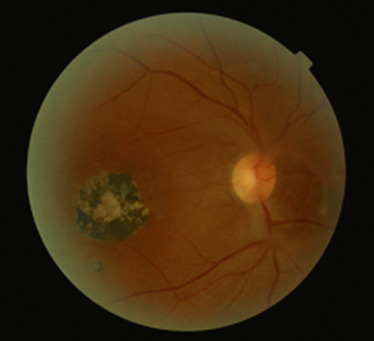
Fig. 14-17 Aspect pseudo-colobomateux d’une cicatrice rétinochoroïdienne au cours d’une toxoplasmose congénitale.
La toxoplasmose oculaire congénitale concerne actuellement 1 enfant pour 10 000 naissances [5]. La séroprévalence de la toxoplasmose parmi les femmes enceintes françaises serait de 50 % , le risque pour une femme non immunisée de s'infecter pendant la grossesse (primo-infection) étant de 0,5 à 1,5 % . Le passage transplacentaire du toxoplasme, et donc l'infection de l'enfant, se produit globalement dans un tiers des cas, dans environ 70 % au cours du dernier trimestre de la grossesse mais dans seulement 5 % au premier trimestre [6]. Les mesures de prévention primaire sont proposées aux femmes enceintes immunocompétentes séronégatives pour la toxoplasmose ou immunodéprimées quel que soit leur statut sérologique (encadré 14-1). En présence d'une primo-infection chez une femme enceinte, il est de principe en France de débuter un traitement par Rovamycine (9 millions d'unités/jour), destiné à prévenir l'infection fœtale. Les conséquences de l'infection sont potentiellement plus graves en cas de contamination précoce avec notamment risque d'anomalies neurologiques, découvertes à l'échographie anténatale ou au cours de la première année de vie. Le risque de lésions oculaires existe pour tous les enfants quelle que soit la date de contamination maternelle. À partir d'une cohorte prospective de 327 enfants atteints de toxoplasmose congénitale, avec un suivi moyen de 14 ans (médiane de 8 ans), une incidence de 24 % de rétinochoroïdites a été notée, avec une récidive dans 29 % des cas [7]. Les pics d'apparition de ces rétinochoroïdites se situaient d'une part entre la naissance et l'âge de 1 an et d'autre part entre 7 et 8 ans. Du point de vue fonctionnel, 69 % des enfants présentaient une acuité visuelle normale, aucun n'ayant de baisse d'acuité visuelle bilatérale. Si la rétinochoroïdite est la manifestation la plus fréquente de la toxoplasmose congénitale, d'autres manifestations ophtalmologiques existent : le strabisme, la microphtalmie, la cataracte, le décollement de rétine, l'atrophie du nerf optique, le nystagmus, le glaucome, l'uvéite antérieure (ou iridocyclite), la néovascularisation choroïdienne et la phtise du globe oculaire [8]. S'il existe une toxoplasmose congénitale symptomatique à la naissance, il faut traiter l'enfant. Néanmoins le bénéfice du traitement en l'absence de symptômes est encore discuté, expliquant l'attitude de certains pays européens à ne pas recommander le dépistage de la toxoplasmose congénitale [9].
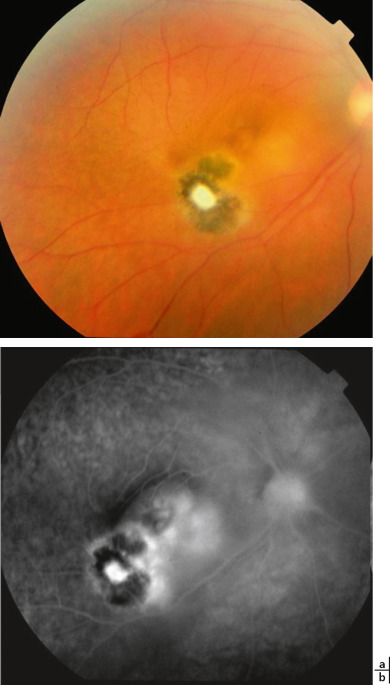
Fig. 14-18 Récidive d’une toxoplasmose oculaire, avec visualisation d’un nouveau foyer de rétinochoroïdite, sur le versant nasal d’une cicatrice pigmentée et atrophique (a).
Le cliché d’angiographie à la fluorescéine (b) met en évidence une hyperfluorescence (centripète) du nouveau foyer de rétinochoroïdite, avec décollement séreux de la rétine neurosensorielle maculaire (en cours de remplissage). La hyalite en regard du pôle postérieur est bien visible.
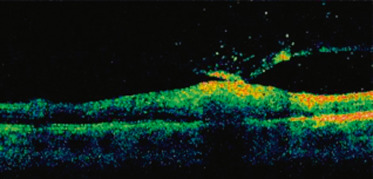
Fig. 14-19 OCT time-domain d’un foyer de rétinochoroïdite active.
L’OCT montre une hyperréflectivité des couches internes de la rétine neurosensorielle, avec en regard des cellules inflammatoires intravitréennes, représentées par des points hyperréflectifs et un décollement du vitré localisé.
La toxoplasmose oculaire acquise est à traiter formellement dans certaines situations (encadré 14-2), même si beaucoup d'équipes la traitent de façon systématique. Le traitement antibiotique permet probablement de réduire le risque de récidive mais il n'y a pas de preuve actuelle qu'il améliore le pronostic visuel, même si l'absence de preuve n'est pas la preuve de l'absence [10]. Différentes molécules thérapeutiques antiparasitaires sont actuellement disponibles pour le traitement « curatif » : la pyriméthamine (Malocide) (hors AMM chez l'enfant), la sulfadiazine (Adiazine) et, hors AMM, la clindamycine (Dalacine), le cotrimoxazole (Bactrim), l'azithromycine (Zithromax) et l'atovaquone (Malarone), pour les principales [11]. La corticothérapie fait partie intégrante de l'arsenal thérapeutique pour la majorité des auteurs et son utilisation rend indispensable la mise en route d'un traitement antiparasitaire afin d'éviter une forme fulminante de toxoplasmose. Classiquement, une bithérapie antiparasitaire est toujours préférée. Le traitement de première intention d'une rétinochoroïdite toxoplasmique peut être le traitement classique de référence ou un traitement modifié « plus moderne » , comme nous l'indiquons dans le , chez l'adulte et chez l'enfant. En effet, les sulfamides présentent des effets secondaires telle la toxidermie allergique de Lyell, potentiellement gravissime, à la différence des macrolides. Le Bactrim (cotrimoxazole = triméthoprime + sulfaméthoxazole), qui correspond ainsi à une bithérapie, peut être utilisé seul et donc constituer une alternative [12]. Le cotrimoxazole permet d'éviter le recours à l'acide folinique, d'où une meilleure observance. Il présente une moindre toxicité hématologique, un coÛt inférieur et existe sous une forme orale liquide pour les enfants. Sa posologie adulte (ou enfant de plus de 15 ans) est Bactrim forte 1 comprimé matin et 1 comprimé soir et sa posologie pédiatrique est de 6 mg/kg/j de triméthoprime et de 30 mg/kg/j de sulfaméthoxazole, en deux prises par jour. En cas d'allergie aux sulfamides, outre l'azithromycine (Zithromax), la clindamycine (Dalacine) per os peut être une alternative, avec un risque néanmoins de colite pseudo-membraneuse. Il est employé chez l'enfant de plus de 15 ans à la dose de 300 à 600 mg/j, en 4 prises et chez l'enfant de plus de 6 ans à 4 à 8 mg/kg/j, en 4 prises également. Deux récentes études prospectives randomisées en simple insu ont montré l'équivalence d'un traitement par injection intravitréenne de 1 mg de clindamycine (0,1 ml) et 400 µg de dexaméthasone au traitement standard par pyriméthamine, sulfadiazine et prednisolone [13,14]. En revanche, pour une efficacité identique des deux protocoles (même réduction de taille, même gain d'acuité visuelle, même réduction de la hyalite, même taux de récidive à 2 ans), les effets secondaires étaient notablement moindres dans le groupe injection intravitréenne. L'efficacité et la sécurité de ce traitement moderne restent à confirmer par une utilisation à plus grande échelle avec un recul supérieur. Néanmoins, cette voie d'administration semble être une option intéressante pour les femmes enceintes ou en cas d'intolérance ou d'allergie au traitement antibiotique systémique [15]. Justement, en présence d'une rétino choroïdite toxoplasmique active chez une femme enceinte, exposant possiblement à un risque de transmission au fœtus, le traitement pourrait être envisagé de façon systématique à base d'azithromycine seule ou, en cas de menace maculaire, associée à la pyriméthamine à partir du 2 trimestre de grossesse, du fait de sa toxicité en début de grossesse [16] : l'injection intravitréenne de clindamycine est une autre option thérapeutique.
Mesures de prévention primaire proposées aux femmes enceintes immunocompétentes séronégatives pour la toxoplasmose ou immunodéprimées
- Ne manger que de la viande bien cuite dans toute son épaisseur et éviter la charcuterie à base de viande crue. Cependant la congélation pendant au moins 72 heures détruit les kystes parasitaires.
- Lors des manipulations de viande crue, de terre ou de légumes souillés de terre, ne pas se toucher la bouche ou les yeux et se laver ensuite soigneusement les mains.
- Laver soigneusement fruits et légumes avant consommation
- Porter des gants pour jardiner.
- Éviter tout contact avec du matériel (bacs, litières, etc.) ayant pu être contaminé par des matières fécales de chat.
- Si l’on possède un chat, il est préférable de ne pas le nourrir de viande crue (préférer les aliments en boîte) et de ne pas s’occuper de sa litière. Si cela est inévitable, porter des gants et les désinfecter à l’eau bouillante
- Ne pas entreposer la litière du chat dans la cuisine.
Indications formelles de traitement curatif en cas de rétinochoroïdite toxoplasmique
- Un foyer menaçant la macula ou la papille optique..
- Une neurorétinite ou papillite
- Une hyalite dense (≥ 1 croix), afin de débuter un traitement corticoïde systémique.
- Une rétinite nécrosante multifocale ou diffuse.
- Des complications de type occlusion vasculaire, décollement de rétine, membrane épirétinienne, néovascularisation choroïdienne, oedème maculaire cystoïde
- Patients immunodéprimés
- Acuité visuelle inférieure à 20/40.
- Un foyer de grande taille (> 2 à 3 diamètres papillaires), quelle que soit sa localisation.
- Une uvéite antérieure sévère.

AMM : autorisation de mise sur le marché ; cp : comprimé ; j : jour ; NFP : numération formule plaquettes ; PNN : polynucléaire neutrophile.
La toxoplasmose oculaire reste une uvéite potentiellement cécitante. Son diagnostic repose davantage sur une présomption clinique que sur des arguments paracliniques, notamment chez l'enfant pour lequel le recours à la ponction de chambre antérieure n'est pas aisé. Elle est à l'origine soit d'une infection congénitale, soit le plus souvent acquise. Elle est traitée systématiquement par de plus en plus d'auteurs, certaines indications thérapeutiques étant consensuellement formelles. L'arsenal thérapeutique s'élargit de plus en plus, avec différentes molécules et voies d'administration, la voie intravitréenne permettant d'éviter les effets indésirables systémiques graves à l'origine de la mauvaise réputation des traitements antiparasitaires de la toxoplasmose.
[1] Henderly DE, Genstler AJ, Smith RE, Rao NA Changing patterns of uveitis Am J Ophthalmol ( 1987 ) :103: 131-136
[2] Scherrer J, Iliev ME, Halberstadt M Visual function in human ocular toxoplasmosis Br J Ophthalmol ( 2007 ) :91: 233-236
[3] Gilbert RE, Stanford MR Is ocular toxoplasmosis caused by prenatal or postnatal infection? Br J Ophthalmol ( 2000 ) :84: 224-226
[4] Bonfioli AA, Orefice F Toxoplasmosis Semin Ophthalmol ( 2005 ) :20: 129-141
[5] Stanford MR, Tan HK, Gilbert RE Toxoplasmic retinochoroiditis presenting in childhood: clinical findings in a UK survey Br J Ophthalmol ( 2006 ) :90: 1464-1467
[6] Dunn D, Wallon M, Peyron F Mother-to-child transmission of toxoplasmosis : risk estimates for clinical counselling Lancet ( 1999 ) :353: 1829-1833
[7] Wallon M, Kodjikian L, Binquet C Long-term ocular prognosis in 327 children with congenital toxoplasmosis Pediatrics ( 2004 ) :113: 1567-1572
[8] Kodjikian L, Wallon M, Fleury J Ocular manifestations in congenital toxoplasmosis Graefes Arch Clin Exp Ophthalmol ( 2006 ) :244: 14-21
[9] Petersen E Toxoplasmosis Semin Fetal Neonatal Med ( 2007 ) :12: 214-223
[10] Pradhan E, Bhandari S, Gilbert RE, Stanford M Antibiotics versus no treatment for toxoplasma retinochoroiditis Cochrane Database Syst Rev ( 2016 ) CD002218
[11] Bodaghi B, Toitou V, Fardeau C Toxoplasmosis : new challenges for an old disease Eye ( 2012 ) :26: 241-244
[12] Soheilian M, Sadoughi M, Ghajarnia M Prospective randomized trial of trimethoprim/sulfamethoxazole versus pyrimethamine and sulfadiazine in the treatment of ocular toxoplasmosis Ophthalmology ( 2005 ) :112: 1876-1882
[13] Soheilian M, Ramezani A, Azimzadeh A Randomized trial of intravitreal clindamycin and dexamethasone versus pyrimethamine, sulfadiazine, and prednisolone in treatment of ocular toxoplasmosis Ophthalmol ( 2011 ) :118: 134-141
[14] Baharivand N, Mahdavifard A, Fouladi RF Intravitreal clindamycin plus dexamethasone versus classic oral therapy in toxoplasmic retinochoroiditis : a prospective randomized clinical trial Int Ophthalmol ( 2013 ) :33: 39-46
[15] Harrell M, Carvounis PE Current treatment of toxoplasma retinochoroiditis : an evidence-based review J Ophthalmol ( 2014 ) :2014: 273506 p
[16] Kodjikian L Toxoplasmose et grossesse J Fr Ophtalmol ( 2010 ) :33: 362-367
M. Boiché, B. Bodaghi, K. Angioi-Duprez
Malgré leur incidence moins élevée que les atteintes antérieures d'origine rhumatismale, les uvéites postérieures ou totales auto-immunes représentent une entité majeure du fait de leur sévérité, l'agressivité du traitement médical et le pronostic visuel potentiellement réservé. Ce sous-chapitre traite les principales entités cliniques composant ce groupe hétérogène d'affections auto-immunes ou auto-inflammatoires.
Maladie de Vogt-Koyanagi-Harada
La maladie de Vogt-Koyanagi-Harada (VKH) est une maladie systémique inflammatoire caractérisée par la survenue d'une panuvéite bilatérale avec présence de multiples décollements séreux rétiniens, associés à des atteintes cutanées (poliose, vitiligo, alopécie), ORL et neurologiques. La physiopathologie exacte du VKH est encore incomplètement élucidée, mais il est désormais communément admis qu'il s'agit d'un processus auto-immun médié par les lymphocytes T et dirigé contre un ou plusieurs antigènes des mélanocytes. Ce mécanisme pourrait être déclenché par une infection à EBV [1].
Cette maladie atteint préférentiellement les femmes entre la troisième et la quatrième décennie, mais peut cependant survenir chez l'enfant [2]. Ainsi, dans une cohorte de 23 enfants atteints de VKH, Abu El-Asrar et al. retrouvent un âge moyen de survenue de la maladie de 12,5 ± 2,4 ans et une large prédominance féminine (87 % ) [3]. Il existe également une prépondérance de la pathologie chez les sujets mélanodermes originaires d'Asie du Sud-Est, du pourtour méditerranéen, d'Amérique centrale et du Sud.
La maladie évolue en quatre phases : phase prodromique, phase uvéitique aiguë, phase de convalescence et phase chronique récurrente. Le diagnostic positif est posé en fonction des critères diagnostiques révisés en 2001 par un comité international (encadré 14-3) [4] et ne peut être évoqué qu'en l'absence de traumatisme ou de chirurgie intra-oculaire préalable, qui orientent d'avantage vers une ophtalmie sympathique, dont le tableau clinique est très proche.
Chez l'enfant, les principaux signes fonctionnels sont une baisse d'acuité visuelle associée à des prodromes à type de céphalées et d'acouphènes [3]. L'examen clinique retrouve une inflammation de chambre antérieure (synéchiante dans environ 30 % des cas) associée à la présence de décollement séreux rétinien et d'un œdème papillaire à l'examen du fond d'œil. La sévérité de la baisse d'acuité visuelle est liée à l'importance et à la localisation des décollements séreux rétiniens et à l'atteinte du nerf optique. L'angiographie à la fluorescéine met en évidence des diffusions en tête d'épingle avec une accumulation progressive de fluorescéine dans les espaces sous-rétiniens. L'angiographie au vert d'indocyanine montre un retard de la perfusion choroïdienne et la présence de granulomes choroïdiens. Quant à l'examen en OCT, il reste un élément indispensable à la mise en évidence des multiples soulèvements rétiniens et à leur suivi [5]. En l'absence de traitement efficace, les décollements séreux rétiniens évoluent en cicatrices choriorétiniennes nummulaires et le fond d'œil prend un aspect dépigmenté dit « en coucher de soleil » . Des signes périphériques comme la poliose et le vitiligo apparaissent durant la phase chronique de la maladie (fig. 14-20).
Le traitement de l'inflammation du segment antérieur consiste en l'instillation de corticoïdes topiques et de collyres cycloplégiques. Le traitement systémique comprend une corticothérapie par voie orale à la dose de 1 mg/kg/j (parfois précédée de l'administration de trois bolus intraveineux) pour une durée minimale de 6 mois [5]. En cas de formes fréquemment récurrentes ou de menace visuelle grave, il peut être proposé un traitement immunosuppresseur. Certains auteurs proposent des immunosuppresseurs à titre systématique mais cela reste controversé.
Fig. 14-20 Vitiligo lors de la phase chronique d’une maladie de Vogt-Koyanagi-Harada.
L'ophtalmie sympathique est une maladie très rare chez l'enfant : l'incidence de sa survenue après une plaie du globe est estimée à 0,24 % [6]. Il s'agit d'une panuvéite auto-immune, bilatérale et granulomateuse survenant au décours d'une plaie transfixiante de l'œil, traumatique (fig. 14-21) ou chirurgicale. Elle est considérée comme une réaction auto-immune médiée par les lymphocytes T et dirigée contre les antigènes des mélanocytes de la choroïde exposés lors de la plaie [7]. Cette hypothèse physiopathologique explique sa grande ressemblance clinique avec la maladie de Vogt-Koyanagi-Harada. En effet, l'ophtalmie sympathique est caractérisée par une uvéite antérieure granulomateuse et synéchiante associée à une hyalite et à des granulomes choroïdiens (fig. 14-22), voire dans les formes sévères à des décollements séreux rétiniens. Les signes extra-oculaires (poliose, vitiligo, céphalées, méningite, perte de l'audition) sont rares au cours d'une ophtalmie sympathique. Le traitement est topique (corticoïdes, mydriatiques), associé à une corticothérapie systémique et souvent à un traitement immunosuppresseur. La gravité des complications justifie cette attitude agressive.
La sarcoïdose est une granulomatose multisystémique rare chez l'enfant. Il existe deux formes distinctes de sarcoïdose en pédiatrie. La première, qui survient chez l'enfant âgé de 10 et 15 ans, est proche de la forme de l'adulte. Elle consiste en une atteinte multiviscérale dans 70 % des cas [8] avec, fréquemment, des adénopathies hilaires ainsi qu'une atteinte pulmonaire, tout comme des symptômes généraux aspécifiques (fièvre, amaigrissement). La seconde, à début précoce, atteint l'enfant de moins de 5 ans et consiste en une triade regroupant éruption cutanée, uvéite et arthropathie [9]. Les frontières avec le syndrome de Blau restent floues dans ce second cas. Le diagnostic de certitude est anatomopathologique par la mise en évidence sur une biopsie d'un granulome épithélioïde et gigantocellulaire sans nécrose caséeuse.
L'uvéite antérieure est la manifestation la plus fréquente de la sarcoïdose chez l'enfant. Elle survient selon les séries chez 24 à 58 % des enfants [10]. Elle se présente sous la forme d'une uvéite synéchiante avec précipités rétrocornéens en graisse de mouton à la partie inférieure de la cornée. Des nodules iriens (de Busacca et de Koeppe) sont fréquemment retrouvés. Il peut exister une Ophtalmie sympathique atteinte postérieure avec choroïdite multifocale et granulomes choroïdiens. Les formes chroniques ou récurrentes peuvent aboutir à des complications comme celles des uvéites antérieures survenant lors d'une AJI : kératopathie en bandelette, hypertonie oculaire, cataracte, œdème maculaire.
Le traitement comprend, outre la corticothérapie topique de l'uvéite antérieure, une corticothérapie systémique à la dose de 1 à 2 mg/kg/j, selon l'âge de l'enfant et la gravité, pendant 4 à 8 semaines, jusqu'à la régression des signes cliniques. Puis, après décroissance progressive, un traitement d'entretien est maintenu pendant au moins 6 mois [10].
Critères diagnostiques de la maladie de Vogt- Koyanagi-Harada [4]
- Absence d’antécédent de traumatisme pénétrant oculaire ou de chirurgie oculaire ayant précédé l’installation des premiers signes d’uvéite.
- Absence d’élément clinique ou biologique évoquant une autre pathologie oculaire.
- Atteinte oculaire bilatérale pour laquelle les critères A ou B doivent être remplis, en fonction du stade de la maladie auquel le patient est examiné :
• Critères A. manifestations précoces de la maladie :
-
choroïdite diffuse pouvant se manifester selon l’une des
manières suivantes :
- accumulation focale de liquide sous-rétinien,
- décollements séreux rétiniens bulleux ;
- dans les cas douteux à l’examen du fond d’oeil, les deux
éléments suivants doivent être présents :
- zones focales de retard de perfusion choroïdienne, multiples zones de fuite en « têtes d’épingle » , larges aires placoïdes d’hyperfluorescence, remplissage en pooling de l’espace sous-rétinien, imprégnation de la papille à l’angiographie à la fluorescéine,
- à l’échographie, épaississement choroïdien diffus, sans signe de sclérite postérieure ;
• Critères B. manifestations tardives de la maladie :
- histoire de la maladie suggérant la présence de l’un des critères précédents et la présence de 2 et 3 ou de multiples signes de 3,
- dépigmentation oculaire (n’importe laquelle des manifestations
ci-dessous est suffisante) :
- fond d’oeil en coucher de soleil (sunset glow fundus),
- signe de Sugiura (vitiligo périlimbique),
- autres signes oculaires (n’importe laquelle des manifestations
ci-dessous est suffisante) :
- cicatrices choriorétiniennes nummulaires dépigmentées,
- altérations et migrations de l’épithélium pigmentaire rétinien,
- uvéite antérieure chronique ou récurrente ;
- manifestations neurologiques et/ou auditives (qui
peuvent avoir disparues au moment de l’examen) (n’importe
laquelle des manifestations ci-dessous est suffisante) :
- syndrome méningé,
- acouphènes
- pléiocytose du liquide céphalorachidien ;
- manifestations cutanées (qui ne peuvent pas avoir précédé
le début de l’atteinte oculaire ou neurologique) (n’importe
laquelle des manifestations ci-dessous est suffisante) :
- alopécie,
- poliose,
- poliose,
Forme complète : critères 1 à 5.
Forme incomplète : critères 1 à 3 et 4 ou 5.
Forme probable, forme oculaire isolée : critères 1 à 3.
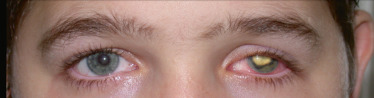
Fig. 14-21 Ophtalmie sympathique de l’oeil droit faisant suite à un traumatisme de l’oeil gauche.
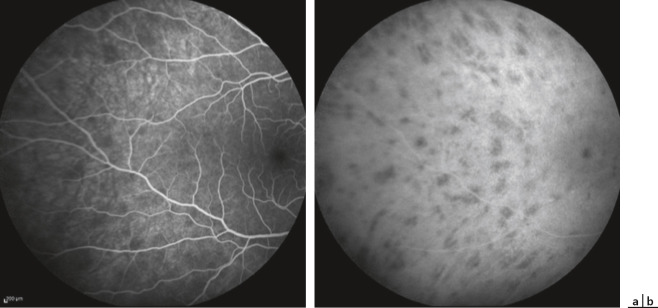
Fig. 14-22 a, b. Analyse angiographique au cours d’une ophtalmie sympathique mettant en évidence des granulomes choroïdiens actifs nécessitant un traitement anti-inflammatoire et immunosuppresseur agressif.
Le syndrome de Blau, ou granulomatose systémique familiale juvénile, est une maladie génétique à transmission autosomique dominante décrite par Edward Blau en 1985 [11]. Cette pathologie monogénique résulte d'une mutation type « gain de fonction » du domaine NBD (nucleotide binding domain) du gène NOD2 [12]. Le syndrome de Blau se rapproche de la sarcoïdose à début précoce par sa présentation clinique en triade associant éruption cutanée, polyarthrite et uvéite. L'atteinte cutanée survient généralement dans la première année de vie sous forme d'une éruption fine maculopapulaire du tronc et des extrémités. L'arthrite est polyarticulaire chez 95 % des patients et touche essentiellement les articulations distales avec classiquement une déformation des articulations interphalangiennes proximales en flexion de type camptodactylie. Elle est associée dans 40 % des cas à une ténosynovite [13].
L'uvéite se déclare chez 80 % des patients, vers l'âge de 4 ans. Bilatérale et synéchiante, l'inflammation antérieure est granulomateuse avec parfois présence de nodules iriens. Une atteinte postérieure potentiellement sévère, caractérisée par une hyalite et une choroïdite multifocale, est décrite dans 75 % des cas. Les complications de type kératopathie en bandelette, cataracte et hypertonie sont fréquentes (plus d'un tiers des cas) dans cette maladie, avec un risque de baisse d'acuité visuelle non négligeable à long terme [12 - 14].
Le traitement comprend la prise en charge topique de l'uvéite antérieure et une corticothérapie systémique. Le recours à des thérapies immunomodulatrices est fréquemment nécessaire. Plusieurs études ont, en ce sens, montré l'intérêt bénéfique des anti-TNF-α et des inhibiteurs de l'IL-1 [15 , 16].
La maladie de Behçet est une vascularite systémique caractérisée par des aphtes buccaux récidivants, des ulcères génitaux, une atteinte inflammatoire oculaire et des lésions cutanées. Les critères diagnostiques de l'International Study Group for Behçet's Disease manquent de sensibilité chez l'enfant et ne peuvent être appliqués à la lettre en pédiatrie, mais la présence d'une aphtose buccale récidivante reste un élément indispensable au diagnostic. Les autres critères de la maladie surviennent avec les années et sont en général retrouvés à l'âge adulte.
L'âge moyen de survenue de l'uvéite est estimé autour de 14 ans, avec une prédominance d'atteinte antérieure chez les moins de 10 ans et une majorité de panuvéite avec vascularite rétinienne chez les grands enfants et les adolescents [17]. Elle survient chez 30 à 60 % des enfants atteints de la maladie de Behçet et est bilatérale dans les deux tiers des cas [18 , 19]. L'atteinte antérieure est toujours non granulomateuse et l'atteinte du segment postérieur associe des lésions nécrotico-hémorragiques à une vascularite (fig. 14-23) qui touche plus fréquemment les veines que les artères avec de possibles complications occlusives. Les atteintes du nerf optique sont principalement dues à l'inflammation ou à l'hypertension intracrânienne sur thrombophlébite cérébrale (fig. 14-24). Le pronostic serait meilleur que chez l'adulte même si cela reste controversé [20]. Les principales complications sont la survenue d'une cataracte et d'une atrophie optique [21].
Selon les recommandations de l'European League Against Rheumatism (EULAR), le traitement de l'atteinte oculaire fait appel aux corticoïdes par voie systémique (et locaux en cas d'atteinte antérieure) associés à l'azathioprine (Imurel) [22]. Les agents biologiques sont nécessaires dans les formes sévères mettant en jeu le pronostic visuel.
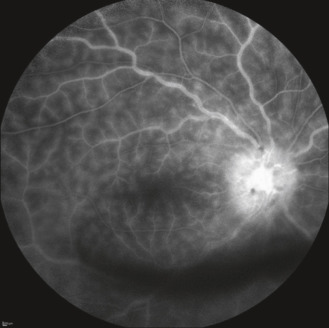
Fig. 14-23 Choriocapillaropathie diffuse et oedème papillaire au cours d’une maladie de Behçet chez un jeune garçon de 8 ans, d’origine antillaise.
La maladie de Kawasaki est une vascularite systémique du jeune enfant affectant les vaisseaux de petit et de moyen calibre. Elle atteint l'enfant de moins de 5 ans et son diagnostic est uniquement clinique, fondé sur l'association d'une fièvre durant plus de 5 jours à au moins quatre des cinq critères suivants : conjonctivite bilatérale, atteinte des muqueuses oropharyngées, exanthème polymorphe, érythème et desquamation des extrémités, adénopathie cervicale de plus de 1,5 cm. La maladie est considérée comme incomplète lorsque seuls deux à trois des critères sont présents. Suivant les séries, une uvéite antérieure aiguë survient chez 13 à 66 % des enfants atteints de maladie de Kawasaki [23 , 24]. L'atteinte est bilatérale, parfois associée à des précipités rétrocornéens fins [25]. Elle survient à la phase aiguë ou subaiguë de la maladie mais régresse sans séquelles en 1 mois [24]. D'autres symptômes ophtalmologiques ont été décrits comme la kératite ponctuée superficielle, l'hémorragie sous-conjonctivale et dans de rares cas un œdème papillaire.
La maladie de Kawasaki atteignant les enfants très jeunes, le seul signe évocateur d'uvéite est généralement l'hyperhémie conjonctivale. Cependant, l'examen ophtalmologique systématique en cas de suspicion de Kawasaki est important sur le plan du diagnostic pédiatrique, car il apporte un argument diagnostique supplémentaire, notamment en cas de forme incomplète; de plus, sur le plan de la prise en charge, il est démontré que l'administration précoce, dans les dix premiers jours de la survenue de la maladie, d'immunoglobulines G par voie intraveineuse diminue significativement le risque d'atteinte des artères coronaires (de plus de 15 % à moins de 5 % ), ce qui abaisse significativement le taux de mortalité des enfants concernés [26].
Le syndrome CINCA ( chronic infantile neurological cutaneous and articular= pathologie chronique infantile neurologique avec atteinte cutanée et articulaire), également appelé syndrome NOMID (neonatal onset multisystem inflammatory disease) est la forme la plus sévère des syndromes CAPS (cryopyrin-associated periodic syndromes), continuum de maladies auto-inflammatoires héréditaires liées à une mutation du gène NLRP3, codant pour la cryopyrine qui régule la formation de l'IL-1. Il existe quelques cas de transmission familiale, mais la maladie est généralement sporadique. Il se manifeste par une triade clinique associant éruption cutanée, atteinte articulaire et manifestations neurologiques. L'éruption est précoce, avant 6 mois de vie, de type urticarien. L'arthrite est sévère, déformante et atteint en particulier les genoux. L'atteinte neurologique est caractérisée par des céphalées et par le développement d'une surdité de perception. L'évolution de la maladie est chronique avec l'installation de poussées de fièvre récurrentes.
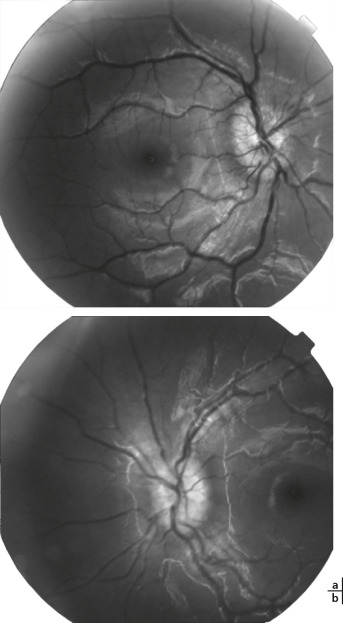
Fig. 14-24 a, b. OEdème papillaire bilatéral compliquant une hypertension intracrânienne par thrombophlébite cérébrale au cours d’une maladie de Behçet.
Les complications ophtalmologiques surviennent vers l'âge de 4 ans. La principale est l'œdème papillaire (fig. 14-25) ou l'atrophie du nerf optique, présent chez plus de 80 % des patients [27, 28]. Près de la moitié des patients ont une inflammation du segment antérieur, non granulomateuse et non synéchiante. Les autres manifestations ophtalmologiques sont un syndrome sec, une rougeur périlimbique et rarement une inflammation du segment postérieur [27]. Des atteintes cornéennes comme une kératopathie en bandelette ou une kératite stromale interstitielle ont été décrites. Le traitement, outre les corticoïdes par voie systémique peu efficaces, repose sur l'utilisation d'un antagoniste du récepteur de l'IL-1, l'anakinra (Kineret), dont l'efficacité a été démontrée [29, 30 ].
Le syndrome d'IRVAN ((idiopathic retinitis vasculitis aneurysms neuroretinis), caractérisé par l'existence d'une vascularite rétinienne, de dilatations anévrismales des artérioles rétiniennes (fig. 14-26) et d'une neurorétinite, est rare chez l'adulte et exceptionnel chez les enfants. La maladie peut rester longtemps asymptomatique. Une baisse d'acuité visuelle est souvent révélatrice. Elle peut être liée à une atteinte maculaire ischémique ou œdémateuse ou à une hémorragie intravitréenne secondaire au développement d'une néovascularisation rétinienne [31]. Parmi les rares cas publiés d'atteinte pédiatrique, le tableau clinique est souvent incomplet : il manque en particulier la composante ischémique qui survient ultérieurement pendant l'évolution de la maladie [32]. Souvent idiopathique, le syndrome d'IRVAN peut être associé à la présence de p-ANCA (perinuclear anti-neutrophil cytoplasmic antibody) [33]. Le traitement comprend la prise en charge de l'ischémie rétinienne et de la néovascularisation quand elle existe. La corticothérapie par voie systémique a été proposée; la place des immunosuppresseurs n'est pas clairement définie.
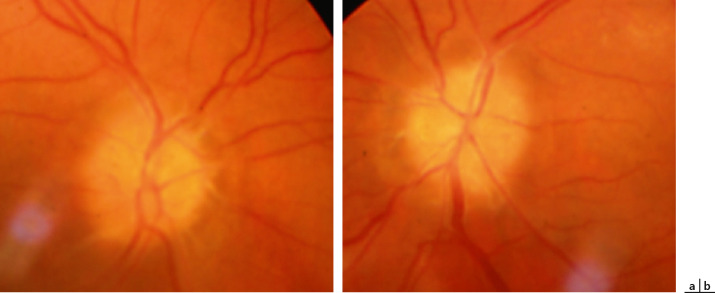
Fig. 14-25 a, b. Altérations papillaires bilatérales au cours d’un syndrome CINCA.
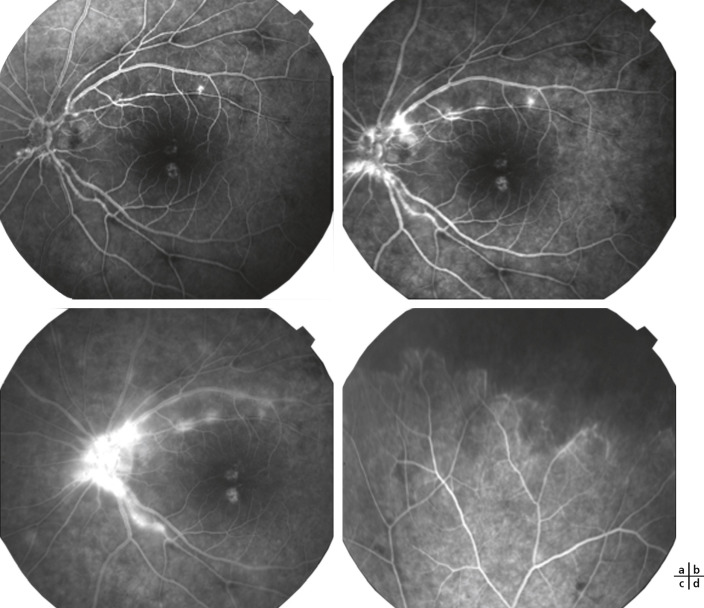
Fig. 14-26 Différents temps angiographiques d’une jeune fille de 13 ans atteinte d’un syndrome d’IRVAN mettant en évidence les macro-anévrismes et l’ischémie rétinienne périphérique.
a. Temps précoce. b. Temps intermédiaire. c. Temps tardif. d. Périphérie.
La granulomatose septique chronique (GSC), maladie génétique rare, est caractérisée par un déficit immunitaire lié à un défaut du métabolisme oxydatif des cellules phagocytaires qui aboutit au développement d'infections bactériennes et fongiques sévères itératives dès la petite enfance. Des atteintes inflammatoires granulomateuses sont associées en particulier aux niveaux pulmonaire, digestif et urinaire [34]. Le diagnostic de la maladie repose sur la capacité à mettre en évidence un déficit du pouvoir oxydatif des phagocytes [34]. Sur le plan ophtalmologique, on retrouve aussi des manifestations de type infectieux (cornée, annexes, segment postérieur) et inflammatoires avec en particulier des granulomes choriorétiniens [35, 36]. Le traitement de la GSC est celui des infections et épisodes inflammatoires et, dans la mesure du possible, sur une prophylaxie au long cours des épisodes infectieux. Le seul traitement curatif disponible à ce jour est l'allogreffe de moelle osseuse dans des situations cliniques précises.
[1] Bassili SS, Peyman GA, Gebhardt BM Detection of Epstein-Barr virus DNA by polymerase chain reaction in the vitreous from a patient with Vogt-Koyanagi-Harada syndrome Retina ( 1996 ) :16: 160-161
[2] Martin TD, Rathinam SR, Cunningham ET Prevalence, clinical characteristics, and causes of vision loss in children with Vogt-Koyanagi-Harada disease in South India Retina ( 2010 ) :30: 1113-1121
[3] Abu El-Asrar AM, Al-Kharashi AS, Aldibhi H Vogt-Koyanagi-Harada disease in children Eye ( 2008 ) :22: 1124-1131
[4] Read RW, Holland GN, Rao NA Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature Am J Ophthalmol ( 2001 ) :131: 647-652
[5] Lavezzo MM, Sakata VM, Morita C Vogt-Koyanagi-Harada disease : review of a rare auto-immune disease targeting antigens of melanocytes Orphanet J Rare Dis ( 2016 ) :24: 11-29
[6] Kumar K, Mathai A, Murthy SI Sympathetic ophthalmia in pediatric age group : clinical features and challenges in management in a tertiary center in southern India Ocul Immunol Inflamm ( 2014 ) :22: 367-372
[7] Kim JB, Jeroudi A, Angeles-Han ST Adalimumab for pediatric sympathetic ophthalmia JAMA Ophthalmol ( 2014 ) :132: 1022-1024
[8] Gedalia A, Khan TA, Shetty AK Childhood sarcoidosis : Louisiana experience Clin Rheumatol ( 2016 ) :35: 1879-1884
[9] Fink CW, Cimaz R Early onset sarcoidosis : not a benign disease J Rheumatol ( 1997 ) :24: 174-177
[10] Shetty AK, Gedalia A Childhood sarcoidosis : a rare but fascinating disorder Pediatric Rheumatology ( 2008 ) :23: 6-16
[11] Blau EB Familial granulomatous arthritis, iritis, and rash J Pediatr ( 1985 ) :107: 689-693
[12] Wouters CH, Maes A, Foley KP Blau syndrome, the prototypic auto-inflammatory granulomatous disease Pediatric Rheumatology ( 2014 ) :12: 33 p
[13] Rose CD, Wouters CH, Meiorin S Pediatric granulomatous arthritis : an international registry Arthritis Rheum ( 2006 ) :54: 3337-3344
[14] Punzi L, Furlan A, Podswiadek M Clinical and genetic aspects of Blau syndrome : A 25-year follow-up of one family and a literature review Autoimmunity Reviews ( 2009 ) :8: 228-232
[15] Milman N, Andersen CB, Hansen A Favorable effect of TNF-a inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation APMIS ( 2006 ) :114: 912-919
[16] Arostegui JI, Arnal C, Merino R NOD2 gene-associated pediatric granulomatous arthritis : clinical diversity, novel and recurrent mutations, and evidence of clinical im- provement with interleukin-1 blockade in a Spanish cohort Arthritis Rheum ( 2007 ) :56: 3805-3813
[17] Sungur GK, Hazirolan D, Yalvac I Clinical and demographic evaluation of Behçet disease among different pediatric age groups Br J Ophthalmol ( 2009 ) :93: 83-87
[18] Atmaca L, Boyvat A, Yalçindağ FN Behçet disease in children Ocul Immunol Inflamm ( 2011 ) :19: 103-107
[19] Koné-Paut I, Yurdakul S, Bahabri SA Clinical features of Behçet's disease in children : an international collaborative study of 86 cases J Pediatr ( 1998 ) :132: 721-725
[20] Kesen MR, Goldstein DA, Tessler HH Uveitis associated pediatric Behçet disease in the American midwest Am J Ophthalmol ( 2008 ) :146: 819-827
[21] Citirik M, Berker N, Songur MS Ocular findings in childhood-onset Behçet disease J AAPOS ( 2009 ) :13: 391-395
[22] Hatemi G, Silman A, Bang D EULAR recommendations for the management of Behçet disease Ann Rheum Dis ( 2008 ) :67: 1656-1662
[23] Burns JC, Joffe L, Sargent RA, Glode MP Anterior uveitis associated with Kawasaki syndrome Pediatr Infect Dis ( 1985 ) :4: 258-261
[24] Alves NR, Magalhães CM, Almeida Rde F Prospective study of Kawasaki disease complications : review of 115 cases Rev Assoc Med Bras ( 2011 ) :57: 295-300
[25] Choi HS, Lee SB, Kwon JH Uveitis as an important ocular sign to help early diagnosis in Kawasaki disease Korean J Pediatr ( 2015 ) :58: 374-379
[26] Newburger JW, Takahashi M, Gerber MA Diagnosis, treatment, and long-term management of Kawasaki disease : a statement for health professionals from the Committee on rheumatic fever, endocarditis, and Kawasaki disease, Council on Cardiovascular Disease in the Young, American Heart Association Pediatrics ( 2004 ) :114: 1708-1733
[27] Dollfus H, Häfner R, Hofmann HM Chronic infantile neurological cutaneous and articular/neonatal onset multisystem inflammatory disease syndrome : ocular manifestations in a recently recognized chronic in- flammatory disease of childhood Arch Ophthalmol ( 2000 ) :118: 1386-1392
[28] Carzoli A, Maalouf T, Henckes O CINCA syndrome : a rare cause of papilledema. The case of homozygous twins J Fr Ophtalmol ( 2010 ) :33: 36-39
[29] Terrada C, Neven B, Boddaert N Ocular modifications in a young girl with cryopyrin-associated periodic syndromes responding to interleukin-1 receptor antagonist anakinra J Ophthal Inflamm Infect ( 2011 ) :1: 133-136
[30] Bascherini V, Granato C, Lopalco G The protean ocular involvement in monogenic autoinflammatory diseases : state of the art Clin Rheumatol ( 2015 ) :3: 1171-1180
[31] Samuel MA, Equi RA, Chang TS Idiopathic retinitis, vasculitis, aneurysms, and neuroretinitis (IRVAN) : new observations and a proposed staging system Ophthalmology ( 2007 ) :114: 1526-1529
[32] Krishnan R, Shah P, Thomas D Subacute idiopathic retinal vasculitis, aneurysms and neuroretinitis (IRVAN) in a child and review of paediatric cases of IRVAN revealing preserved capillary perfusion as a more common feature Eye ( 2015 ) :25: 145-151
[33] Nourinia R, Montahai T, Amoohashemi N Idiopathic retinal vasculitis, aneurysms and neuroretinitis syndrome associated with positive perinuclear antineutrophil cytoplasmic antibody J Ophthalmic Vis Res ( 2011 ) :6: 330-333
[34] Winkelstein JA, Marino MC, Johnston RB Chronic granulomatous disease. Report on a national registry of 368 patients Medicine (Baltimore) ( 2000 ) :79: 155-169
[35] Angioi K, Terrada C, Locatelli A Ocular manifestations of X-linked chronic granulomatous disease : about two atypical case reports Ocul Immunol Inflamm ( 2015 ) :23: 458-461
[36] Locatelli A, Bene MC, Zuily S, Angioi-Duprez K Ocular manifestations in chronic granulomatous disease J Fr Ophtalmol ( 2013 ) :36: 789-795
A.Darugar, B.Bodaghi
Les uvéites postérieures infectieuses non toxoplasmiques sont plus rares chez l'enfant. Cependant, leur méconnaissance conduirait à des complications irréversibles et une mise en jeu définitive du pronostic visuel.
Les rétinites virales sont exceptionnelles chez l'enfant immunocompétent [1]. Chez l'enfant immunodéprimé, les rétinites à CMV sont les plus fréquentes. Elles surviennent lorsque les taux de CD4 sont inférieurs à 50 éléments/mm chez les patients infectés par le VIH. On observe également des rétinites à CMV dans un contexte de déficit immunitaire congénital ou iatrogène post-chimiothérapie ou après greffe de moelle osseuse. L'atteinte est volontiers bilatérale et asymptomatique sur œil blanc. Le vitré est paradoxalement clair. On observe des foyers de rétinite nécrotico-hémorragiques de contenu inhomogène mais à bords assez nets à proximité d'un vaisseau [2]. Le traitement repose sur l'administration intraveineuse d'un traitement antiviral. Les injections intravitréennes sont impossibles à réaliser chez l'enfant sans anesthésie. La restauration immunitaire permet de contrôler l'extension des plages de rétinite et les récidives. Le pronostic visuel est réservé et dépend de la localisation des lésions et de leur extension. Une atrophie optique n'est pas rare.
Les rétinites à CMV congénitales sont liées à une transmission de la mère à l'enfant pendant la grossesse et plus fréquemment durant l'accouchement ou l'allaitement [3].
Les formes nécrotiques rétiniennes de l'enfant sont rares mais particulièrement graves. Le déficit immunitaire est exceptionnel mais certaines formes génétiques ciblées ne sont pas encore identifiées. Les principaux virus responsables sont les membres de la famille herpès. Les études fondées sur l'analyse de l'humeur aqueuse ou du vitré mettent en évidence principalement l'HSV-2 avec une transmission lors de l'accouchement et le passage de la filière génitale. L'encéphalite associée n'est pas exceptionnelle. La présentation clinique est celle d'une panuvéite granulomateuse en général unilatérale avec baisse d'acuité visuelle de degré variable, une uvéite antérieure granulomateuse parfois hypertensive mais peu synéchiante associée à une hyalite dense, une nécrose périphérique évoluant de façon centripète et circonférentielle. Les vasculites associées sont fréquentes ainsi que l'inflammation du nerf optique. Les atteintes plus centrales sont possibles mais rares (fig. 14-27). Le diagnostic formel repose sur l'analyse des liquides oculaires à l'aide des techniques moléculaires. Le traitement doit être très rapidement mis en œuvre reposant sur les antiviraux visant les virus herpès avec adaptation secondaire à l'identification moléculaire. Dans les formes à risque, les dérivés de l'aciclovir (Zovirax® et Zelitrex®) sont abandonnés au profit du ganciclovir (Cymevan®) ou du foscarnet (Foscarvir®). Les injections intravitréennes complémentaires de ganciclovir sont parfois nécessaires. La durée du traitement n'est pas standardisée mais reste prolongée. En effet, même si les récidives sont rares du côté concerné, elles peuvent survenir au niveau de l'œil adelphe, soit concomitamment (bilateral acute retinal necrosis [BARN]), soit après un intervalle libre de 6 semaines à plusieurs dizaines d'années. Le décollement de rétine et l'atrophie optique restent les complications les plus redoutables. Les infections par les virus de la rougeole, de la dengue, du chikungunya, West Nile Ebola et Zika sont des agents potentiellement aussi redoutables au plan rétinien que les virus herpès et il faudra les suspecter en fonction des antécédents des enfants (en particulier les voyages récents). Malheureusement, les solutions thérapeutiques sont quasi inexistantes pour ces infections virales émergentes.
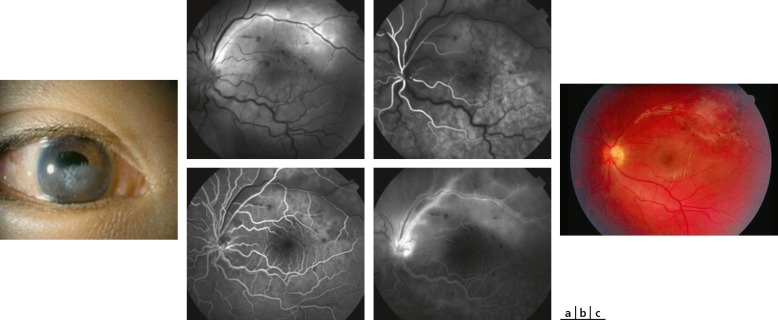
Fig. 14-27 Rétinite nécrosante à HSV-2 chez une jeune adolescente de 14 ans ayant déjà perdu l’oeil droit à 8 ans (a).
b. L’angiographie met en évidence une atteinte nécrotique au niveau de l’arcade temporale supérieure avec décollement séreux rétinien associé. c. Cicatrisation de la nécrose sous traitement antiviral agressif avec récupération visuelle totale.
La maladie des griffes du chat est transmise par une bactérie Bartonella henselae après griffures de chat. L'inflammation intra-oculaire s'installe après plusieurs jours avec une atteinte principalement postérieure de type neurorétinite semblable à la forme décrite par Leber. Elle associe un œdème papillaire, un décollement séreux rétinien et un œdème maculaire avec exsudats en étoile périmaculaire (fig. 14-28). Des foyers sous forme de taches blanches localisées autour de la papille ou plus diffuses pourraient être associés ou exister en dehors de la neurorétinite classique. Le diagnostic repose sur l'interrogatoire des parents, l'examen du fond d'œil et la sérologie. On retrouve parfois une adénopathie dans le territoire d'inoculation. Le traitement consiste en une antibiothérapie systémique : azithromycine (Zithromax), trimétho-prime-sulfaméthoxazole (Bactrim), rifampicine (Rifadine) selon la forme clinique et l'avis du pédiatre ou de l'infectiologue. Les fluroquinolones sont déconseillées chez les enfants en raison des risques ostéo-articulaires. Les cyclines seraient peu efficaces et contre-indiquées chez l'enfant avant 8 ans à cause du risque dentaire. L'intérêt des corticoïdes est très discuté mais il pourrait être proposé en cas d'atteinte sévère centrale.
Sur le plan visuel, l'évolution est favorable sous traitement. L'œdème papillaire régresse au bout de quelques semaines sans laisser d'atrophie optique, les exsudats persistent plus longtemps. La normalisation du fond d'œil s'observe au bout de 6 à 12 mois.
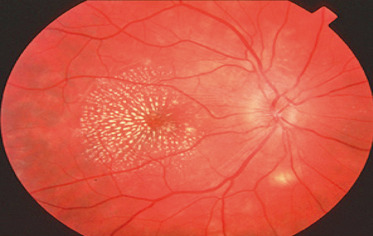
Fig. 14-28 Aspect typique de neurorétinite associée à une maladie des griffes du chat.
La syphilis est une maladie bactérienne due à Treponema pallidum. Il s'agit d'une infection sexuellement transmissible, sa présence chez l'enfant relève principalement de la transmission de la mère à l'enfant pendant la grossesse ou pendant l'accouchement, la transmission post-natale étant exceptionnelle. Aucun cas de transmission par le lait maternel n'a été rapporté.
La syphilis congénitale peut se manifester soit de façon précoce (avant l'âge de 2 ans) avec un risque de mortalité de 20 % , soit de façon tardive après l'âge de 2 ans. L'atteinte oculaire s'intègre dans un cortège de manifestations extra-ophtalmologiques : cutanées et articulaires, hépatomégalie, dentaires et neurologiques.
Sur le plan oculaire, l'atteinte du segment postérieur est prédominante avec un aspect de choriorétinite « poivre et sel » montrant une alternance de plages d'atrophie de l'épithélium pigmentaire et de lésions d'hyperplasie de l'épithélium pigmentaire [4]. Un tableau de rétinite pigmentaire avec atrophie optique et aspect grêle des artérioles rétiniennes peut s'observer de façon exceptionnelle.
Le diagnostic de la syphilis congénitale est clinique et sérologique. Le traitement est systémique avec des injections de pénicilline G par voie intraveineuse. L'efficacité du traitement est suivie par la décroissance puis la négativation des taux sérologiques de VDRL (venereal disease research laboratory). Le pronostic visuel est réservé avec des lésions souvent découvertes au stade cicatriciel.
Il est à noter qu'il existe une atteinte très évocatrice de la syphilis congénitale : la kératite interstitielle de nature probablement immunitaire qui ne répond pas au traitement antibiotique mais cortico-sensible. La triade de Hutchinson est l'association de malformations dentaires (dents de Hutchinson, petites et d'un gris sale), de lésions oculaires (kératite interstitielle bilatérale) et de troubles auditifs (surdité labyrinthique), observée au cours de la syphilis congénitale.
DUSN [5] est l'acronyme de diffuse unilateral subacute neuroretinitis ou neurorétinite subaiguë diffuse unilatérale. Il s'agit d'une réaction inflammatoire choriorétinienne liée à la présence d'un parasite intra-oculaire. Le pathogène en cause n'est pas exactement connu mais plusieurs nématodes ont été identifiés comme Toxocara canis,Baylisacaris procyonis ,Ancylostoma caninum ,Gnathostoma spinigerum ,Strongyloides stercoralis et Brugia malayi.
Le DUSN se manifeste par une baisse d'acuité visuelle, des myodésopsies ou un scotome d'évolution progressive mais extensif. Comme la toxocarose, l'atteinte est strictement unilatérale. L'examen clinique retrouve une hyalite souvent modérée et un aspect de choroïdite diffuse avec des lésions granulomateuses choroïdiennes d'âges différents, une atrophie de l'épithélium pigmentaire, une papillite et une vasculite rétinienne (fig. 14-29).
Le parasite peut être identifié à l'examen minutieux de la rétine lorsque celle-ci n'est pas encore trop remaniée. Les lésions sont plus étendues que celles de la toxocarose (fig. 14-30) (voir plus haut chapitre 14.2 ).
Le traitement consiste en une antibiothérapie antiparasitaire associée à une destruction du nématode au laser lorsque cela est possible.
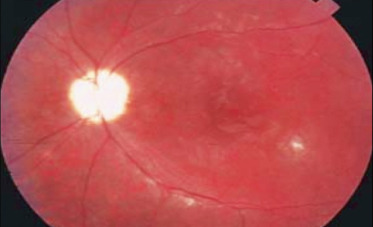
Fig. 14-29 Atrophie rétinienne totale due à un neurorétinite subaiguë diffuse unilatérale (DUSN).
(Remerciements au Pr J. Davis.)
L'uvéite est l'atteinte la plus fréquente de la tuberculose oculaire. Il s'agit le plus souvent d'une atteinte par hypersensibilité de type 4 et rarement d'une dissémination par voie hématogène du bacille de la tuberculose
L'uvéite par hypersensibilité est généralement granulomateuse, très synéchiante voire en secclusion pupillaire avec hypertonie oculaire multifactorielle. Elle peut être compliquée d'une atteinte du segment postérieur non spécifique : œdème papillaire, œdème maculaire. L'atteinte peut être uni- comme bilatérale. L'uvéite peut être associée à une sclérite.
L'uvéite liée à une maladie tuberculeuse active et bacillifère est le plus souvent asymptomatique et se manifeste quant à elle par une atteinte choroïdienne dont la lésion caractéristique est le tubercule de Bouchut. Il s'agit d'un ou de plusieurs granulomes choroïdiens souvent proches du pôle postérieur et parfois associés à une lame de décollement séreux rétinien.
Le diagnostic de l'étiologie tuberculeuse repose sur un faisceau d'argument avec le terrain du patient, la radiographie du thorax, les résultats de l'intradermoréaction (IDR) à la tuberculine et les tests de prolifération lymphocytaire (TB-Spot ou Quantiferon) [6]. Chez l'enfant, le diagnostic est plus difficile à établir : absence de signes cliniques et radiologiques spécifiques, bactériologie souvent peu contributive. L'IDR n'apporte qu'une aide partielle (difficulté de l'injection intradermique chez l'enfant, manque de sensibilité et de spécificité). Les tests de type Quantiferon sont difficiles à interpréter chez les enfants avant l'âge de 5 ans.
Le traitement d'une infection latente repose préférentiellement sur une bithérapie par isoniazide (Rimifon) et rifampicine (Rfadine) pendant 3 mois, comme chez l'adulte.
En l'absence d'atteinte cérébroméningée associée, un schéma classique de quadrithérapie initiale pendant 2 mois puis de bithérapie pendant les 4 mois suivants semble suffisant chez l'adulte dans les formes actives. Chez l'enfant, il est toutefois habituellement recommandé de prolonger le traitement sur une durée totale de 9 mois, soit 2 mois de quadrithérapie et 7 mois de bithérapie. Si une localisation cérébroméningée est associée, un traitement de 12 mois est recommandé.
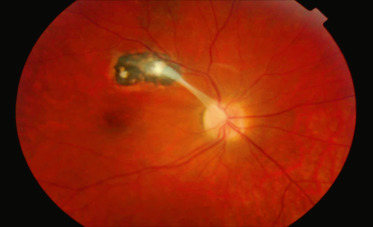
Fig. 14-30 Toxocarose oculaire se manifestant par une cicatrice pigmentée au niveau de l’arcade temporale supérieure et traction vitréorétinienne.
Les uvéites postérieures d'origine infectieuse sont rares chez l'enfant en dehors de la toxoplasmose. Leur prise en charge nécessite un environnement hospitalier multidisciplinaire habitué aux spécificités de l'enfant. L'usage de la corticothérapie est discuté au cas par cas en fonction de la sévérité de l'inflammation associée et des complications.
[1] Gupta A, Rani PK, Bagga B Bilateral herpes simplex-2 acute retinal necrosis with encephalitis in premature twins J AAPOS ( 2010 ) :14: 541-543
[2] Baumal CR, Levin AV, Read SE Cytomegalovirus retinitis in immunosuppressed children Am J Ophthalmol ( 1999 ) :127: 550-558
[3] Alford CA, Stagno S, Pass RF, Britt WJ Congenital and perinatal cytomegalovirus infections Rev Infect Dis ( 1990 ) :12: S745-S753
[4] Friedenwald JS Ocular lesions in fetal syphilis Trans Am Ophthalmol Soc ( 1929 ) :27: 203-218
[5] Gass JD, Scelfo R Diffuse unilateral subacute neuroretinitis J R Soc Med ( 1978 ) :71: 95-111
[6] Bodaghi B, LeHoang P Ocular tuberculosis Curr Opin Ophthalmol ( 2000 ) :11: 443-448
B. Bodaghi , P. LeHoang
L'uvéite pédiatrique n'a pas de présentation clinique spécifique. Il est donc capital d'éliminer les étiologies tumorale, malformative ou génétique prenant le masque d'une inflammation intra-oculaire [1]. L'absence d'analyse minutieuse provoquerait un retard diagnostic significatif avec, par conséquent, la mise en jeu du pronostic visuel ou vital de l'enfant.
Les uvéites pédiatriques représentent approximativement 10 % de l'ensemble des inflammations intra-oculaires. Sur l'ensemble des cas, 5 % pourraient être des fausses uvéites avec des causes tumorales impliquées pour moitié. Une étude récente réalisée au National Institute of Health permet de vérifier ces données, en tout cas pour ce qui concerne les pseudo-uvéites néoplasiques [2].
Le rétinoblastome infiltrant diffus est particulièrement difficile à différencier d'une authentique uvéite à hypopion (fig. 14-31).
Shields et al. ont rapporté les caractéristiques d'une série de 1507 rétinoblastomes, parmi lesquels 32 étaient des formes infiltrantes diffuses, dont deux bilatérales [3]. L'âge moyen au diagnostic de ces formes infiltrantes diffuses était de 4 ans (1,5 à 16 ans); ces enfants étaient adressés pour rétinoblastome dans 76 % des cas mais également pour uvéite (3,9 % ), maladie de Coats (1,3 % ), traumatisme (1,3 % ) et rétinopathie non spécifique (1,3 % ); la biomicroscopie a mis en évidence des dépôts tumoraux au niveau de l'endothélium cornéen (24 % ), un œdème stromal (3,9 % ), un pseudo-hypopion (32 % ), un hyphéma (9 % ), une néovascularisation irienne (50 % ), des nodules tumoraux iriens (18 % ); le segment postérieur était le siège d'une lésion rétinienne infiltrante associée à un ensemencement vitréen (91 % ) et une hémorragie intravitréenne (24 % ); des calcifications ont été diagnostiquées à l'échographie en mode B dans 79 % des cas et en tomodensitométrie dans 89 % des cas. La ponction de chambre antérieure à visée diagnostique reste contre-indiquée sous peine d'ensemencement orbitaire. Une énucléation a dÛ être réalisée dans les 34 yeux, et on ne retrouvait pas de métastase chez ces patients après 47 mois de suivi. Des alternatives thérapeutiques comme la chimiothérapie locale sont en cours d'évaluation dans certaines formes.
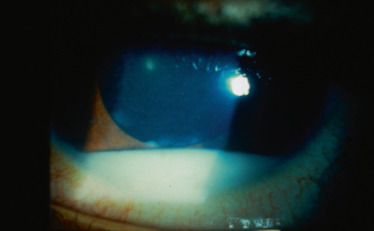
Fig. 14-31 Hypopion à convexité supérieure au cours d’un rétinoblastome infiltrant diffus.
Les atteintes oculaires au cours des leucémies infantiles sont importantes à connaître car elles peuvent révéler l'hémopathie. Les mécanismes physiopathologiques sont multiples : invasion directe par les cellules malignes, rétinopathie liée aux anomalies des différentes lignées hématopoïétiques, neuropathie optique secondaire à une atteinte cérébrale et infections opportunistes. Ainsi, il peut exister une infiltration irienne (fig. 14-32) ou un pseudo-hypopion. Une atteinte choroïdienne peut être responsable de décollement séreux rétinien. Une étude rétrospective a été menée entre 2005 et 2014, portant sur 185 enfants atteints de leucémie et ayant bénéficié d'un examen ophtalmologique [4]; l'âge médian était de 6 ans (0,5 à 18 ans) et le suivi moyen était de 36 mois (0,5 à 108 mois); l'atteinte oculaire était présente dans 24,3 % des cas, parmi lesquels 37,8 % étaient symptomatiques; la prévalence était de 20,4 % au cours des leucémies aiguës lymphocytaires et de 36,4 % au cours des leucémies aiguës myélocytaires (p = 0,0051); le taux de mortalité était plus élevé chez ces dernières mais l'infiltration oculaire n'était pas un facteur de risque; une thrombopénie était plus significativement associée en cas de symptomatologie oculaire.
Fig. 14-32 Infiltration irienne au cours d’une leucémie chez l’enfant.
Histiocytose non langerhansienne, atteignant principalement la peau, elle ne touche l'œil que chez 0,3 à 0,5 % des patients. L'hyphéma (fig. 14-33) avec un œil rouge douloureux et photophobe est une manifestation fréquente et un motif de consultation. L'atteinte cutanée associée peut manquer dans plus de 40 % des cas. Le risque d'atteinte oculaire serait plus élevé chez les enfants de moins de 2 ans atteints de xanthogranulomes multiples. Deux consultations ophtalmologiques par an jusqu'à l'âge de 2 ans sont recommandées dans ce cas. Les autres présentations cliniques sont : les tumeurs de l'iris, le glaucome unilatéral spontané, la pseudo-uvéite et l'hétérochromie irienne. Les paupières ou plus rarement le segment postérieur et l'orbite peuvent aussi être touchés. Une association avec la neurofibromatose de type 1 est possible. Les corticoïdes topiques ou systémiques sont initialement proposés. Le traitement des formes avancées repose sur la chirurgie, la chimiothérapie ou la radiothérapie, et plus récemment par des injections d'anti-vascular endothelial growth factor (anti-VEGF). Le diagnostic différentiel avec le mélanome est possible dans des formes agressives [5, 6].
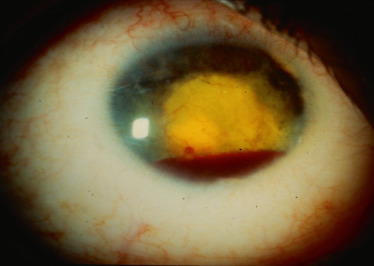
Fig. 14-33 Hyphéma associé à des altérations iriennes au cours d’un xanthogranulome juvénile.
Il s'agit d'une dystrophie rétinienne, décrite pour la première fois par Criswick et Schepens en 1969, responsable d'un arrêt de la vascularisation de la rétine périphérique (fig. 14-34). Les modes de transmission sont multiples : autosomique dominant, récessif lié à l'X ou autosomique récessif. Les mutations du gène frizzled-4 (FZD4) localisé en 11q13-q23 représentent 20 % des formes autosomiques dominantes. Les formes autosomiques dominantes sont aussi associées avec le gène LRP5 situé en 11q13.4 et le locus 11p13-p12. Les formes liées au chromosome X sont causées par des mutations dans le gène de la maladie de Norrie (en Xp11.4).
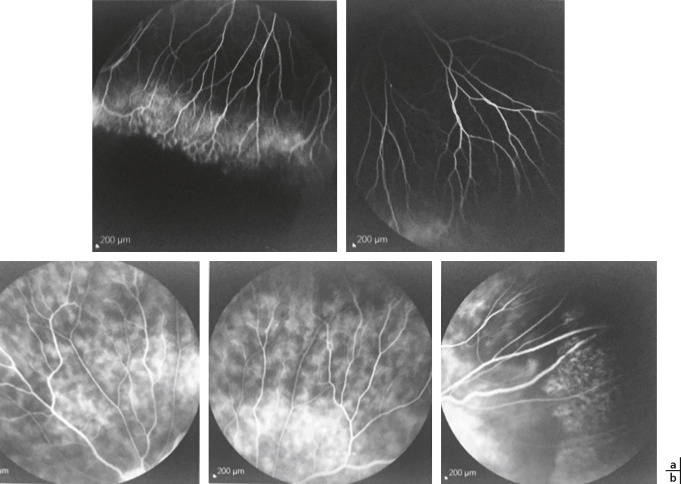
Fig. 14-34 Vitréorétinopathie exsudative familiale.
a. OEil droit : aspect caractéristique d’altération vasculaire périphérique. b. OEil gauche : choriocapillaropathie diffuse évoquant à tort une uvéite intermédiaire.
L'uvéite intermédiaire est l'un des diagnostics différentiels avec atteinte vitréenne et diffusions vasculaires rétiniennes angiographiques. Cependant, l'examen de la périphérie met en évidence des anastomoses artérioveineuses et des proliférations néovasculaires en bordure de l'ischémie ainsi qu'une masse fibrovasculaire temporale périphérique, avec configuration particulière des vaisseaux rétiniens qui sont étirés vers la zone temporale et qui forment un angle aigu en sortant de la papille. Malgré une sévérité hétérogène, les hémorragies intravitréennes, l'exsudation lipidique intrarétinienne, l'œdème et l'ectopie maculaire, la rétraction du vitré entraînant la formation d'un pli rétinien et le décollement de rétine sont les principales complications [7]. Le traitement repose sur la destruction des zones d'ischémie par photocoagulation au laser ou plus rarement une cryoapplication. Les anti-VEGF pourraient être proposés en cas de néovascularisation associée. La chirurgie est indiquée en cas de décollement de rétine.
La prévalence est de 1 sur 3 500 personnes. Les gènes responsables sont très nombreux. La hyalite modérée et l'œdème maculaire sont possibles dans d'authentiques cas de rétinite pigmentaire. Les formes évoluées d'uvéites peuvent également prendre la présentation d'une rétinite pigmentaire. Dans les deux cas, la présence d'ostéoblastes, les altérations électrophysiologiques et le rétrécissement du champ visuel permettent de poser le diagnostic qui sera précisé par l'analyse génétique.
Affection idiopathique, il s'agit de télangiectasies rétiniennes responsables de dépôts d'exsudats intra- ou sous-rétiniens (fig. 14-35). L'atteinte est unilatérale sans anomalie systémique associée et touche les jeunes garçons dans 80 % des cas. La gêne fonctionnelle peut ne pas être au premier plan, car l'enfant ne se plaint pas forcément d'une baisse d'acuité visuelle unilatérale. L'anomalie du reflet pupillaire, la découverte fortuite d'une baisse d'acuité visuelle et/ou l'apparition d'un strabisme peuvent constituer les circonstances de diagnostic. L'examen mettra en évidence des anomalies vasculaires périphériques responsables de l'exsudation avec, à un stade plus tardif, une leucocorie et un décollement de rétine. Les principaux diagnostics différentiels incluent le rétinoblastome, la vitréorétinopathie exsudative familiale, la maladie de von Hippel-Lindau, l'uvéite intermédiaire et l'incontinentia pigmenti. Le traitement repose sur la photocoagulation des lésions vasculaires périphériques lorsqu'elles sont exsudatives et plus rarement la cryoapplication. Le décollement de rétine nécessite une prise en charge appropriée. Le pronostic visuel final des formes sévères demeure réservé [8].
Fig. 14-35 Altérations vasculaires périphériques avec exsudats sous-rétiniens au cours d’une maladie de Coats.
Il s'agit d'une lésion congénitale solitaire, typiquement localisée en temporal de la macula (fig. 14-36), de découverte fortuite sans retentissement visuel [9] mais dont la toxoplasmose pourrait être le diagnostic différentiel. L'hypertrophie congénitale de l'épithélium pigmentaire de la rétine et le syndrome de Gardner sont d'autres affections à considérer. L'atteinte correspond à une anomalie de l'épithélium pigmentaire de la rétine. Les formes bilatérales sont possibles mais rares [10]. L'OCT et l'analyse de l'autofluorescence aident au diagnostic [11]. Aucun traitement n'est nécessaire.
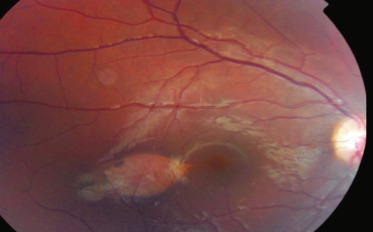
Fig. 14-36 Aspect caractéristique d’une maculopathie en torpille.
[1] Madigan WP, Raymond WR, Wroblewski KJ A review of pediatric uveitis : Part I. Infectious causes and the masquerade syndromes J Pediatr Ophthalmol Strabismus ( 2008 ) :45: 140-149
[2] Grange LK, Kouchouk A, Dalal MD Neoplastic masquerade syndromes in patients with uveitis Am J Ophthalmol ( 2014 ) :157: 526-531
[3] Shields CL, Ghassemi F, Tuncer S Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes Ophthalmology ( 2008 ) :115: 2253-2258
[4] Bitirgen G, Belviranli S, Caliskan U Ophthalmic manifestations in recently diagnosed childhood leukemia Eur J Ophthalmol ( 2016 ) :26: 88-91
[5] Fontanilla FA, Edward DP, Wong M Juvenile xanthogranuloma masquerading as melanoma J AAPOS ( 2009 ) :13: 515-518
[6] Rao A, Padhy D The child with spontaneous recurrent bleeding in the eye BMJ Case Rep ( 2014 ) :2014: pii: bcr2014203925
[7] Gilmour DF Familial exudative vitreoretinopathy and related retinopathies Eye (Lond) ( 2015 ) :29: 1-14
[8] Grosso A, Pellegrini M, Cereda MG Pearls and pitfalls in diagnosis and management of coats disease Retina ( 2015 ) :35: 614-623
[9] Golchet PR, Jampol LM, Mathura JR, Daily MJ Torpedo maculopathy Br J Ophthalmol ( 2010 ) :94: 302-306
[10] Richez F, Gueudry J, Brasseur G, Muraine M Maculopathie en torpille bilatérale J Fr Ophtalmol ( 2010 ) :33: 296 p
[11] Thomas AS, Flaxel CJ, Pennesi ME Spectral-domain optical coherence tomography and fundus autofluorescence evaluation of torpedo maculopathy J Pediatr Ophthalmol Strabismus ( 2015 ) :52: e8-10 Online
D. Guindolet, E. Gabison
L'inflammation de l'épisclère et/ou celle de la sclère sont des affections rares chez l'enfant. Leur incidence est respectivement évaluée à 7,63 et 0,763 cas pour 100 000 habitants et par an, chez les enfants de moins de 14 ans [1]. Chez l'enfant, l'épidémiologie, les caractéristiques cliniques et la prise en charge thérapeutique sont peu décrites dans la littérature contrairement aux formes de l'adulte. L'analyse et la classification cliniques sont cependant identiques à celles de l'adulte, même si certaines étiologies et mesures de prise en charge thérapeutique sont néanmoins spécifiques de l'enfant.
La sclère est une tunique blanche, opaque et externe formant les quatre cinquièmes externes du globe oculaire. Avec une épaisseur variant entre 0,3 et 1 mm, la sclère assure la rigidité de l'œil et permet l'insertion des muscles oculomoteurs. Elle se prolonge en avant par la cornée et en arrière par le nerf optique au niveau du foramen postérieur.
La sclère est composée de trois couches :
- – l'épisclère à l'extérieur, qui est un tissu conjonctif lâche composé de collagène, de protéoglycanes et de fibroblastes;
- – le stroma composé d'un tissu conjonctif dense avec des faisceaux de fibrilles de collagène entrelacées mais aussi de fibres élastiques et de rares sclérocytes;
- – la lamina fusca à la face interne, composée de fibres élastiques et de mélanocytes; cette couche est en contact avec l'uvée.
La nutrition de la sclère est assurée par diffusion à partir du plexus épiscléral, superficiel et profond, et de la choroïde. L'innervation de la sclère est assurée par les nerfs ciliaires courts et longs.
La consultation initiale en cas de sclérite/épisclérite est souvent réalisée dans le cadre de l'urgence, motivée par des douleurs, pouvant être insomniantes en cas de sclérite, associées à une rougeur oculaire. La localisation et l'extension de la rougeur oculaire doivent être appréciées à l'œil nu à la lumière du jour; cet examen permet également d'observer les modifications de la coloration sclérale pouvant témoigner d'épisodes antérieurs. La rougeur est classiquement décrite comme « vive » en cas d'épisclérite et de plus « sombre » voire « violacée » en cas de sclérite. L'examen à la lampe à fente détermine si les vaisseaux dilatés ont une orientation radiaire (aspect « peigné » ), par rapport au limbe dans le cas d'une atteinte du plexus épiscléral superficiel, ou une orientation transverse, entrecroisée (« en bataille » ) dans le cas d'une atteinte du plexus épiscléral profond. L'examen des vaisseaux peut être sensibilisé par l'utilisation d'une lumière verte associée à un filtre anérythre. Une zone d'ischémie doit être recherchée ainsi que la présence d'un nodule scléral. L'examen en fente fine des lignes de profil en regard de la rougeur peut aider à localiser l'inflammation; les lignes du profil antérieur et postérieur sont séparées en regard d'une zone d'épisclérite alors qu'elles restent parallèles, accolées mais refoulées vers l'avant en regard d'une zone de sclérite. Un test de vasoconstriction à la phényléphrine à 10 % (sympathomimétique alpha) fait disparaître la rougeur associée à une épisclérite alors qu'elle persistera dans le cas d'une sclérite. L'utilisation de ce produit est contre-indiquée avant l'âge de 12 ans (avec également une recommandation pour un usage plutôt après 18 ans), mais est cependant couramment employée dans cette indication, en respectant les recommandations d'occlusion des points lacrymaux par compression digitale. Une autre option pourrait être l'utilisation de phényléphrine à 5 % , également contre-indiquée avant 12 ans ou à 2,5 % avant 12 ans, même si l'efficacité du test thérapeutique à ces doses n'est pas précisée. La recherche d'une ulcération conjonctivale ou cornéenne peut être objectivée par l'instillation d'une goutte de fluorescéine et l'utilisation d'une lumière bleu cobalt. La surface oculaire doit être analysée à la recherche de signes de rosacée oculaire, ainsi que le segment antérieur et postérieur à la recherche d'une uvéite associée et de signes évoquant une sclérite postérieure. L'angiographie du segment antérieur est peu utilisée en pratique courante chez l'adulte ou chez l'enfant.
La sclérite postérieure peut être isolée et peut ainsi survenir sur un œil blanc ou, a contrario, être associée à une sclérite antérieure. Le diagnostic doit être évoqué en présence de plis choroïdiens ou rétiniens, d'un décollement de rétine exsudatif ou d'un œdème papillaire. L'OCT peut être utile pour objectiver les plis et la présence de liquide sous-rétinien. L'angiographie à la fluorescéine de la rétine (non systématique) recherche une vascularite, un décollement séreux sous-rétinien, une papillite. L'échographie oculaire en mode B recherche le signe du « T » traduisant un épaississement de la paroi sclérale. En IRM ou en tomodensitométrie (TDM), un épaississement postérieur de la paroi oculaire est recherché. Ces examens sont utiles pour éliminer un diagnostic différentiel (infection, rétinoblastome [2], etc.).
La classification clinique des épisclérites et sclérites de l'enfant est la même que celle utilisée chez l'adulte et repose sur la description faite par Watson, Hayreh et Awdry [3] en 1968. Une épisclérite simple ou nodulaire est liée à une inflammation du plexus épiscléral superficiel. Une sclérite est liée à une inflammation du plexus profond; on distingue les formes antérieure et postérieure de sclérite selon le siège de l'inflammation respectivement en avant ou en arrière de l'ora serrata. Parmi les sclérites antérieures, on distingue les formes diffuses, nodulaires et nécrosantes. Aucun cas de scleromalacia perforans n'a été décrit chez l'enfant.
La distinction des différentes entités d'épisclérite et de sclérite joue un rôle déterminant du point de vue pronostique et thérapeutique chez l'adulte. Chez l'enfant, cette nosologie est beaucoup moins claire compte tenu de la rareté des descriptions dans la littérature; ainsi le raisonnement réalisé chez l'adulte est ici transposé chez l'enfant.
Les pathologies pouvant induire une épisclérite récidivante ou un épisode de sclérite sont listées dans l'encadré 14-4 ; les manifestations oculaires peuvent être inaugurales d'une pathologie inflammatoire générale à ne pas méconnaître. Le diagnostic de la pathologie causale associe un examen clinique général ainsi qu'un bilan paraclinique. Chez l'enfant, une prise en charge conjointe avec un pédiatre ou un médecin interniste est souhaitable afin de cibler les explorations paracliniques à visée diagnostique, plus difficiles chez l'enfant et potentiellement invasives, mais également pour assurer le suivi des thérapeutiques et leurs potentiels effets secondaires; par ailleurs, certaines pathologies inflammatoires pédiatriques ne disposent pas de marqueurs spécifiques et reposent sur un faisceau d'arguments cliniques. L'épisode de sclérite peut être isolé et les signes extra-ophtalmologiques peuvent apparaître secondairement; un suivi prolongé est nécessaire afin d'assurer une prise en charge précoce.
Pathologies responsables d’une inflammation sclérale
- Arthrite juvénile idiopathique (AJI) :
- forme à début systémique (FS-AJI ou maladie de Still) ;
- forme à début oligoarticulaire ou polyarticulaire ;
- enthésites en rapport avec une arthropathie (ERA) ;
- rhumatisme psoriasique.
- Connectivites
- lupus érythémateux systémique (LES) ;
- dermatomyosites juvéniles ;
- sclérodermie de l’enfant.
- Vascularites :
- artérite de Takayasu ;
- péri-artérite noueuse de l’enfant ;
- maladie de Kawasaki ;
- granulomatose avec polyangéite ;
- syndrome de Churg-Strauss ;
- polychondrite atrophiante ;
- cryoglobulinémie ;
- vascularite de Cogan ;
- syndrome de Behçet.
- Sarcoïdose.
- Entérocolopathies :
- maladie de Crohn ;
- rectocolite hémorragique.
- Vascularite à IgA.
- Syndrome de Goodpasture.
- Rhumatisme articulaire aigu.
- Porphyrie congénitale.
- Fièvre périodique (syndrome de Marshall).
- Fièvre méditerranéenne familiale.
- Thyroïdite de Hashimoto.
- Granulomatose infectieuse (tuberculose, lèpre, syphilis, maladie de Lyme).
- Infection :
- virale (herpès simplex, zona, hépatites B et C, cytomégalovirus) ;
- bactérienne et fongique.
- Surgically induced necrotizing scleritis (SINS).
- Iatrogène : post-chirurgie (strabisme, ptérygion).
- Bisphosphonate.
- Idiopathique.
Une attention particulière doit être portée lors de l'interrogatoire à la recherche d'infections à HSV ou VZV. La présence de sclérite antérieure unilatérale souvent nodulaire et présentant une ulcération conjonctivale en regard doit systématiquement faire évoquer le diagnostic. La présence de vésicules conjonctivales au cours de l'éruption varicelleuse est à la frontière du diagnostic différentiel, car elle peut s'accompagner d'une réaction inflammatoire épisclérale superficielle ou plus profonde et secondairement faire le lit d'une atteinte sclérale plus inflammatoire qu'infectieuse.
D'autres pathologies infectieuses peuvent se révéler par des sclérites ou des épisclérites. Les conjonctivites bactériennes sévères sont ainsi fréquemment associées à une inflammation épisclérale, indiquant une antibiothérapie locale. D'autres infections heureusement plus rares comme la tuberculose peuvent également être révélées par une atteinte sclérale souvent nodulaire pouvant être à prédominance infectieuse ou inflammatoire.
Le contexte récent ou même ancien d'une chirurgie oculaire doit conduire à un examen minutieux à la recherche de l'extrusion de fils de strabisme (fil résorbable de Vicryl en cas de chirurgie récente ou fil de Nylon d'une myopexie ancienne) ou de matériel d'indentation d'un décollement de rétine opéré ab externo.
Les atteintes oculaires allergiques des kératoconjonctivites vernales, comme celles des kératoconjonctivites phlycténulaires associées aux rosacées de l'enfant, sont plus souvent compliquées d'épisclérites que de sclérites. Dans ce dernier cas, l'atteinte est fréquemment à prédominance unilatérale et répond particulièrement bien aux anti-inflammatoires locaux.
Il n'existe pas de recommandations spécifiques pour le traitement des sclérites de l'enfant. La plupart des cas de sclérite antérieure sont inclus dans les cohortes de patients d'âge adulte et ne permettent pas de différencier les thérapeutiques utilisées pour cette population. Seules les descriptions dédiées aux cas de sclérite postérieure de l'enfant semblent indiquer le recours fréquent aux corticoïdes oraux et aux immunosuppresseurs pour contrôler l'inflammation. Par ailleurs, l'évolution récente de la nosologie et de la connaissance en termes de physiopathologie, ainsi que le développement de biothérapies ciblées ont profondément modifié la prise en charge des pathologies inflammatoires de l'enfant. Les biothérapies permettent de limiter la durée et les doses requises de corticoïdes oraux et ainsi d'en limiter les effets indésirables.
Leur utilisation n'est pas recommandée [4]. Ils ne doivent pas être utilisés en cas d'altération de la surface oculaire (risque de kératolyse).
Les AINS par voie orale sont indiqués en cas de sclérite antérieure diffuse ou nodulaire, seuls ou en association avec des corticoïdes locaux. Leur utilisation doit cependant être évitée en cas d'entérocolopathie (risque de poussée inflammatoire, perforation digestive) ou d'insuffisance rénale, par exemple dans le cadre d'une glomérulonéphrite. L'échec de ce traitement doit faire proposer un recours aux corticoïdes systémiques.
Les AIS topiques sont à utiliser en association avec les AINS en cas d'épisclérite ou de sclérite antérieure diffuse ou nodulaire. Ils pourraient avoir un rôle d'épargne cortisonique en association au traitement systémique en cas de sclérite [5].
Les corticoïdes oraux doivent être proposés en cas d'échec des AINS ou en première intention pour les cas de sclérite antérieure sévère ou postérieure. Le traitement est instauré à une dose initiale est de 1 mg/kg/j [6] et suivi d'une décroissance progressive sur 4 à 6 semaines. Les effets secondaires des corticoïdes systémiques pourraient être limités par leur utilisation en prise par alternance un jour sur deux. Une cortico-résistance ou une cortico-dépendance doivent motiver l'introduction d'un traitement immunosuppresseur.
La réalisation répétée de bolus de 10 à 15 mg/kg/j peut être proposée pour les cas de sclérite antérieure avec atteinte cornéenne, de sclérite nécrosante ou de sclérite postérieure. Le nombre d'injection est guidé par la réponse au traitement. Un relais par corticoïdes oraux doit être réalisé, éventuellement en association avec un immunosuppresseur.
La réalisation d'une injection sous-conjonctivale de dexaméthasone voire sous-ténonienne de triamcinolone est difficile dans un contexte pédiatrique sans anesthésie, si ce n'est chez certains adolescents. L'efficacité de ces injections en association au traitement systémique a été rapportée dans un cas de sclérite antérieure diffuse non nécrosante chez un enfant [7].
Le recours à des immunosuppresseurs est nécessaire en cas de sclérite cortico-résistante, cortico-dépendante ou en cas d'association à une pathologie systémique le justifiant. La plupart des cas décrits de sclérite postérieure semble indiquer la nécessité d'un recours aux immunosuppresseurs en première intention en association avec les corticoïdes oraux. Le choix du type d'immunosuppresseur est guidé par la pathologie sous-jacente. Les antimétabolites (méthotrexate, azathioprine, mycophénolate mofétil) ainsi que la ciclosporine sont les traitements les plus anciens et les plus décrits dans la littérature. Considérant les effets secondaires du cyclophosphamide et l'existence d'alternatives (biothérapie), leur utilisation chez l'enfant est à éviter. Les biothérapies sont de plus en plus employées chez l'enfant pour la prise en charge des pathologies inflammatoires systémiques. Plusieurs cas de sclérites postérieures de l'enfant ont été traités avec succès par anti-TNF-α (infliximab [8]) ou antirécepteur à l'IL-6 (tocilizumab [9]). Le choix du type de biothérapie est guidé par la pathologie systémique à l'origine de la sclérite.
[1] Honik G, Wong IG, Gritz DC Incidence and prevalence of episcleritis and scleritis in Northern California Cornea ( 2013 ) :32: 1562-1566
[2] Shenoy R, Suryawanshi M, Isaac R, Philip SK Posterior scleritis in pediatric age group : a case report and review of literature Oman J Ophthalmol ( 2016 ) :9: 59-62
[3] Watson PG, Hayreh SS, Awdry PN Episcleritis and scleritis. I Br J Ophthalmol ( 1968 ) :52: 278-279 contd
[4] Williams CPR, Browning AC, Sleep TJ A randomised, double-blind trial of topical ketorolac vs artificial tears for the treatment of episcleritis Eye Lond Engl ( 2005 ) :19: 739-742
[5] McMullen M, Kovarik G, Hodge WG Use of topical steroid therapy in the management of nonnecrotizing anterior scleritis Can J Ophthalmol ( 1999 ) :34: 217-221
[6] Cheung CMG, Chee SP Posterior scleritis in children : clinical features and treatment Ophthalmology ( 2012 ) :119: 59-65
[7] Sen HN, Ursea R, Nussenblatt RB, Buggage RR Subconjunctival corticosteroid injection for the treatment of non-necrotising anterior scleritis Br J Ophthalmol ( 2005 ) :89: 917-918
[8] Weiss K, Rieger R, Keitzer R, Pleyer U Successful infliximab treatment of posterior scleritis in a 13-year-old child refractory to other immunosuppressive therapy Graefes Arch Clin Exp Ophthalmol ( 2007 ) :245: 1735-1737
[9] Loricera J, Blanco R, Hernández JL Tocilizumab in patients with Takayasu arteritis : a retrospective study and literature review Clin Exp Rheumatol ( 2016 ) :34: S44-S53