Pathologies du cristallin chez l'enfant
Coordonné par C. Speeg-Schatz
C. Speeg-Schatz, D. Thouvenin
Les cataractes chez l'enfant sont plus rares que chez l'adulte mais représentent chez l'enfant la première cause de leucocorie et de pathologie cristallinienne [1-3]. La plupart des cataractes sont malformatives et congénitales (elles existent à la naissance ou sont d'apparition retardée; les formes plus tardives sont souvent traumatiques ou plus rarement métaboliques).
Le caractère obturant importe, car il entrave le développement visuel, ainsi que l'âge d'installation qui conditionne le pronostic fonctionnel.
Ainsi en fonction de l'âge d'opacification, on distingue :
les cataractes congénitales, uni- ou bilatérales, opaques dès la naissance. Lorsque l’opacité est obturante, elle est grave en termes d’amblyopie. Quelques cataractes, dont la pyramidale antérieure, sont non obturantes et non évolutives et de fait de meilleur pronostic ;
les cataractes infantiles, qui s’opacifient dans les deux premières années de vie, lors du pic de la période sensible du développement visuel (6 mois à 2,5 ans) ;
les cataractes juvéniles qui s’opacifient avant 10 ans. Leur pronostic est d’autant meilleur que l’opacification est tardive ;
les cataractes traumatiques qui exposent aux mêmes complications que chez l’adulte auxquelles se rajoutent les risques et problèmes thérapeutiques de l’amblyopie.
Les cataractes précoces bilatérales représentent une cause importante de cécité dans le monde chez l'enfant, exposant à une malvoyance si la prise en charge est tardive; c'est une cause de cécité évitable.
Les cataractes unilatérales posent le problème des malformations oculaires souvent associées (microphtalmie, persistance hyperplasique du vitré primitif, etc.) et de l'amblyopie unilatérale.
Toute cataracte obturante requiet un traitement chirurgical et un traitement bien codifié de l'amblyopie.
Les pathologies systémiques pouvant être aussi responsables de cataracte chez l'enfant ont des causes qui surviennent pendant la période prénatale (causes génétiques et/ou infectieuses, pathologies de la grossesse, expositions à des toxiques in utero); elles sont les plus fréquentes avec particulièrement celles d'origine génétique, qui d'ailleurs sont responsables de 30 à 50 % de l'ensemble des cas de déficits visuels durant l'enfance [4].
En termes de prévalence, les causes génétiques représentent environ 0,19 à 0,44 ‰ enfants [5] et sont plus fréquentes chez le garçon, en raison des maladies héréditaires liées à l'X (sex-ratio de 1,5 à comparer au sex-ratio de 1,3 pour les étiologies non génétiques) [6]. Globalement, la prévalence des causes prénatales varie de 0,43 à 0,76 ‰ enfants selon les études [5, 7-12].
La prévalence des étiologies péri- et néonatales (asphyxie à la naissance, autres complications périnatales, prématurité) est de l'ordre de 0,20 à 0,30 ‰ enfants [5, 7-10, 12]; elle est significativement plus élevée dans le sexe masculin en raison d'une morbidité périnatale plus fréquente chez le garçon.
Les pathologies de la période juvénile (infections, malnutrition, accidents) ont joué un grand rôle, en termes de prévalence de la cataracte chez l'enfant, dans le passé et encore aujourd'hui dans les pays en voie de développement. En revanche, elles sont actuellement en recul dans les pays « industrialisés » où elles concernent 0,03 à 0,12 ‰ enfants [5, 7-12].
Les anomalies congénitales de l'œil (cataracte, microphtalmie, anophtalmie, colobome de l'iris, glaucome, mégalocornée, aniridie, etc.) sont souvent pourvoyeuses de déficiences visuelles sévères; toutefois, elles représentent des malformations d'une relative rareté, Une étude européenne collaborative, EUROCAT, portant sur 1 832 857 naissances, a permis de préciser leur prévalence dans la population générale : elle varie selon la malformation entre 2,3 et 14 pour 10 000 naissances, avec une prévalence globale évaluée à 6 pour 10 000 [13]. Ces chiffres sont comparables aux données françaises publiées par le registre des malformations congénitales du Bas-Rhin portant sur 131 760 grossesses consécutives dans le département : la prévalence s'y élève à 5,9 pour 10 000 naissances pour les générations 1979 à 1988 [14].
Prévalence : d'après Foster et al, [15], on peut estimer la prévalence de la cataracte congénitale entre 1 et 15 cas pour 10 000 enfants et celle de la cécité (acuité visuelle ou AV < 3/60) liée à la cataracte de l'ordre de 0,1 à 0,4 cas pour 10 000 enfants dans les pays occidentaux, La prévalence des cataractes congénitales bilatérales varie de 1 à 5 pour 10 000 naissances selon les auteurs, En France, Stoll et al, [14] ont établi un taux de prévalence des cataractes congénitales à 2,3 pour 10 000 pour les générations 1979 à 1988.
Prévalence dans les populations à risque : certains enfants présentent des risques élevés de développer une pathologie de la vision dans leur enfance tels que les nouveau-nés de faible âge gestationnel ou de faible poids de naissance, les enfants ayant présenté une anoxie périnatale ou une infection anté- ou néonatale (toxoplasmose, infections virales, notamment à cytomégalovirus), les enfants avec antécédents familiaux ou porteurs d'anomalies chromosomiques (trisomie 21, par exemple).
L'ensemble des études publiées dans la littérature internationale souligne l'importance de faire un examen systématique avec dilatation et fond d'œil chez ces enfants, dès les premiers mois de vie et régulièrement ensuite, afin de détecter le plus précocement possible des anomalies organiques qui perturbent la fonction visuelle et peuvent affecter le développement de ces enfants [16].
-
Cataracte polaire antérieure (fig. 13-1) : elle est souvent bilatérale, héréditaire avec un retentissement visuel minime et en règle générale non évolutives; elle est souvent observée comme un petit point blanc dans l’aire pupillaire, il s’agit d’un défaut de migration de la vésicule cristallinienne; il convient de la surveiller, car elle est souvent associée à un astigmatisme lui-même amblyogène,
-
Cataracte pyramidale antérieure (fig. 13-2) : l’opacité pointe en chambre antérieure et elle est déjà plus amblyogène car volontiers associée à une sous-capsulaire antérieure qui a tendance à s’étendre et à devenir obturante,
-
Cataracte sous-capsulaire antérieure : souvent acquise, elle est souvent post-traumatique ou secondaire à une inflammation ou s’intègre dans un contexte d’atopie ou un syndrome d’Alport.
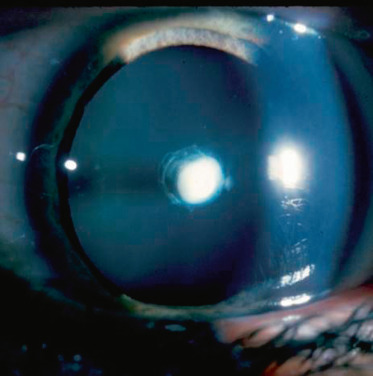
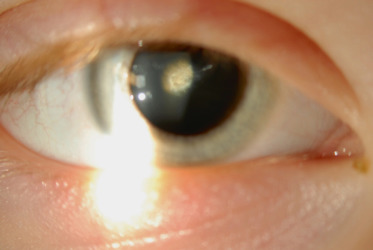
Fig. 13-2 Cataracte pyramidale antérieure.
Il existe une opacification évolutive du noyau fœtal. On parle de cataracte fœtale. Au départ, la périphérie du cristallin reste claire; le plus souvent bilatérale, parfois asymétrique, elle n’est pas toujours du ressort chirurgical; elle peut s’intégrer dans un tableau malformatif de microcornée et/ou de microphtalmie, ou dans une trisomie 21. Les cataractes nucléaires ou centrales peuvent être :
-
céruléennes (fig. 13-5) : faite d’opacités arrondies, ponctuées, situées dans les couches superficielles du noyau fœtal et adulte donc de développement tardif;
-
poussiéreuses centrales (fig. 13-6);
-
nucléaires fœtales (fig. 13-7);
-
zonulaires (ou lamellaires) (fig. 13-8) : c’est l’opacification progressive des couches entourant le noyau; on retrouve un aspect d’arceau, à cheval autour du noyau et souvent bilatéral et héréditaire; elles peuvent être nucléaires, avec ou sans cavaliers périphériques, corticales ou suturales, mais peuvent avoir des aspects très variables;
-
stellaires, coralliformes (fig. 13-9).
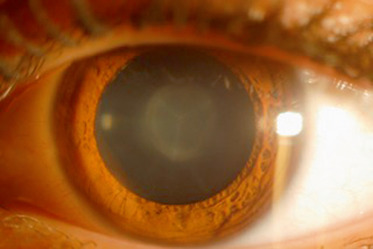
Fig. 13-3 Cataracte suturale et nucléaire.

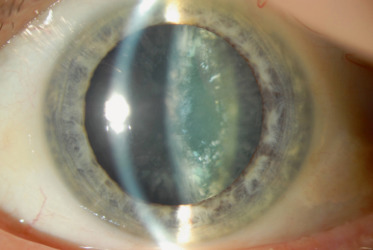
Fig. 13-5 Cataracte céruléenne.
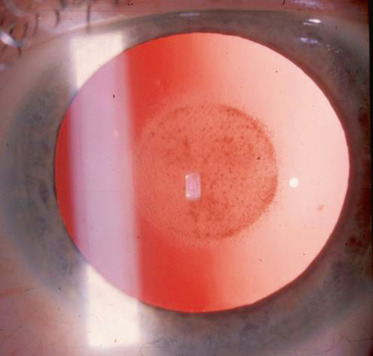

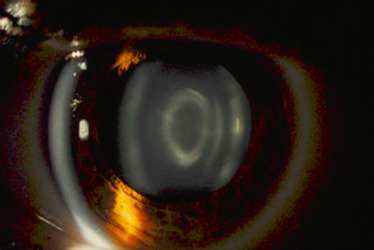
Fig. 13-8 Cataracte zonulaire.
-
Cataracte polaire postérieure : le plus souvent unilatérale, isolée et non héréditaire, découverte lors d’un bilan d’amblyopie et/ou de strabisme, dont le lenticône postérieur en est une variante (fig. 13-10).
-
Lenticône postérieur (fig. 13-11) : c’est une protrusion conique vers l’arrière du cristallin, le plus souvent isolée et unilatérale, mais qui peut être bilatérale. Cela donne l’aspect au fond d’œil d’une « goutte d’huile » . Elle induit une myopie cristallinienne et en général une amblyopie profonde. Cette forme doit faire rechercher un syndrome d’Alport.
-
Cataracte sous-capsulaire postérieure : souvent acquise ou post-traumatique fréquemment, elle peut aussi être idiopathique, post-irradiation ou secondaire à une corticothérapie prolongée. Elle donne une baisse d’acuité et une photophobie.

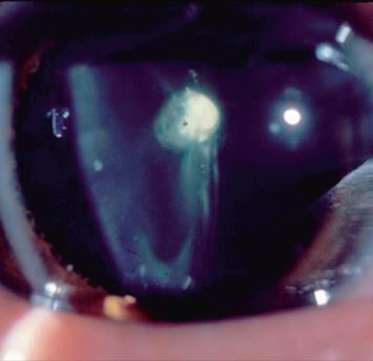
Fig. 13-11 Lenticône postérieur.
Stade ultime de l’évolution de la cataracte : tout le cristallin est blanc laiteux donnant une franche leucocorie. Elles sont parfois partiellement régressives.
-
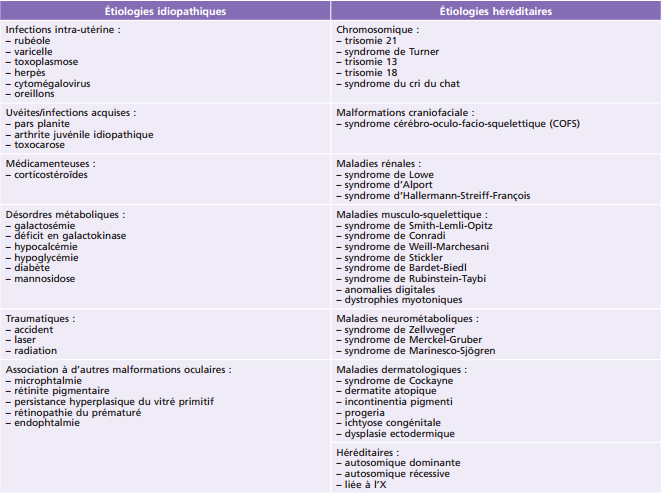
Persistance de la vascularisation fœtale : le plus souvent unilatérale par résorption incomplète du vitré primitif, avec souvent étirement des procès ciliaires. La microphtalmie est quasi constante.
-
Les formes bilatérales sont souvent malformatives, il en existe plusieurs :
-
tache de Mittendorf et persistance de l’artère hyaloïdienne. Petite tache blanche sur la capsule postérieure, souvent excentrée en nasal, cicatrice d’une régression incomplète de l’artère hyaloïdienne (cordon avasculaire tendu vers la papille). Souvent associée à une amétropie et une amblyopie (fig. 13-12a-d);
-
persistance de la membrane pupillaire : reliquats de la tunica vasculosa lentis, parfois associée à un ectropion de l’uvée;
-
persistance de la tunique vasculaire postérieure du cristallin donnant une opacité rétrocristallinienne blanche vascularisée et étirement des procès ciliaires;
-
papille de Bergmeister : saillie d’un tissu de soutien entourant l’artère hyaloïde ou voile avasculaire devant la papille. Elle est asymptomatique et non évolutive.
-
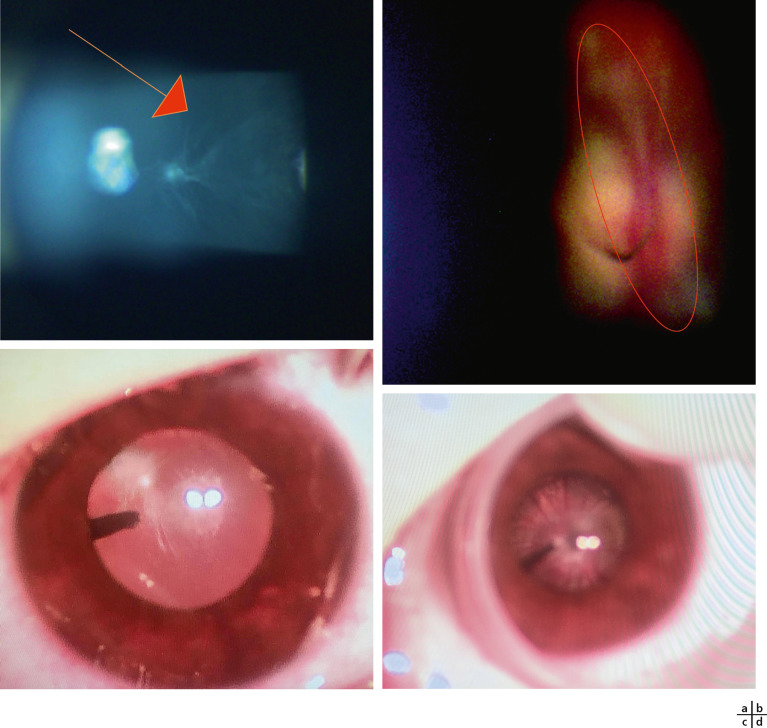
Fig. 13-12 Persistance de l’artère hyaloïdienne.
a. Tache de Mittendorf en nasal à disposition étoilée et radiaire. b. Visualisation du canal de Cloquet en se défocalisant dans le vitré. c. Canal de Cloquet. d. Tache de Mittendorf. (Remerciements au Pr D. Denis.)
Les causes les plus fréquentes sont les infections intra-utérines, les maladies ou désordres métaboliques et les syndromes génétiques (voir tableau 13-1).
Les formes unilatérales sont souvent malformatives et sporadiques.
Chez l'enfant en bonne santé apparente un bilan étiologique préopératoire n'est pas nécessaire en dehors des sérologies habituelles (toxoplasmose, oreillons, rubéole, rougeole, cytomégalovirus, herpès, etc.), d'un caryotype et d'une échographie cardiaque (pour éliminer d'éventuelles malformations associées). Ainsi le bilan sera orienté par l'aspect de la cataracte, l'examen général de l'enfant et celui de sa famille.
Toute cataracte congénitale devra faire l'objet d'une consultation génétique.
Nous distinguerons les cataractes précoces congénitales des cataractes tardives acquises :
cataractes congénitales : nous verrons les cataractes héréditaires, métaboliques, secondaires à une infection foetomaternelle, syndromique ou idiopathique ;
cataractes acquises : la plupart sont traumatiques, ou secondaires à une pathologie oculaire (uvéites, iridocyclite hétérochromique de Fuchs, pathologie du segment postérieur, etc.) ou générales (myotonie de Steinert, Kearns-Sayre, rétinopathie pigmentaire, diabète, etc.), ou encore iatrogènes (corticothérapie prolongée, irradiation).
Ainsi on peut considérer qu'un tiers des cataractes est héréditaire, un tiers syndromique ou entrant dans une autre pathologie, un tiers idiopathique. En pratique, on ne trouvera une cause que dans environ la moitié des cas.
Tableau 13-1 Étiologies des cataractes chez l’enfant.
Dans une forme bilatérale de cataracte, avant de retenir le diagnostic de cataracte idiopathique, il faut être certain que les parents sont indemnes de toute atteinte cristallinienne afin d’éliminer une forme héréditaire autosomique dominante et avoir recours à un avis pédiatrique afin d’éliminer une cause systémique.
De même, devant une cataracte unilatérale et isolée dont le bilan général est négatif, on retiendra ce diagnostic d’exclusion de cataracte idiopathique
Dans les cataractes héréditaires familiales, toutes les formes de transmission ont été décrites mais la transmission autosomique dominante est la plus fréquente (75 % des cas). Elles sont variables d’un membre de la famille à l’autre, en règle bilatérales, souvent nucléaires et zonulaires. S’il s’agit d’un premier cas de cataracte bilatérale héréditaire, un bilan pédiatrique est indispensable afin d’éliminer un syndrome héréditaire dont il s’agirait du premier cas : syndrome de Lowe (oculo-cérébro-rénal, lié à l’X), syndrome d’Alport (autosomique dominant, avec néphropathie, surdité, en rapport avec une anomalie du collagène), syndrome de Bardet-Biedel (petite taille, obésité, retard mental, polydactylie, hypogonadisme, rétinopathie pigmentaire, rares cataractes); il faut également dans ces cas réaliser un caryotype pour éliminer une trisomie 21.
Congénitales ou de développement plus tardif, totales ou partielles, les cataractes chromosomiques s’observent volontiers dans la trisomie 21; beaucoup plus rarement dans les trisomies 13, 15, 18 et de la délétion partielle du bras court du chromosome 5 ou syndrome du cri du chat
On peut les observer dans l’oxycéphalie, le syndrome de Crouzon ou d’Apert (craniosténoses) ou encore dans la dyscéphalie en tête d’oiseau ou syndrome de Hallermann-Streiff-François (aplasie du maxillaire inférieur, nez mince, anomalies dentaires, nanisme, hypotrichose avec alopécie, atrophie cutanée sur le visage, microphtalmie et cataracte bilatérale). Parmi les anomalies squelettiques, nous citerons les syndromes de : Smith-Lemli-Opitz, Conradi, Weill-Marchesani, Stickler, Bardet-Biedel, Rubinstein-Taybi.
Dystrophie myotonique de Steinert.
-
Syndrome oculo-cérébro-rénal de Lowe : hérédité liée à l’X. Dysfonctionnement rénal tubulaire (hypophosphatémie, hyperphosphaturie, acidose tubulaire et hyperamino-acidurie), glaucome et cataracte, hypotonie et bosses frontales proéminentes, retard mental.Syndrome oculo-cérébro-rénal de Lowe : hérédité liée à l’X. Dysfonctionnement rénal tubulaire (hypophosphatémie, hyperphosphaturie, acidose tubulaire et hyperamino-acidurie), glaucome et cataracte, hypotonie et bosses frontales proéminentes, retard mental.
-
Syndrome d’Alport : néphropathie hématurique, surdité de perception, cataracte en lenticône antérieur.
Les cataractes peuvent aussi être associées à des anomalies cutanées (ichtyose héréditaire liée à l’X, syndrome de Rothmund-Thomson, etc.), osseuses (maladie des épiphyses pointillées) ou encore cérébrales (syndrome de Sjögren associant cataracte et oligophrénie, syndrome de Sotos, gigantisme cérébral ou syndrome de Marinesco-Sjögren associant syndrome cérébelleux, cataracte et retard psychomoteur).
Dominées par la galactosémie avec cataracte en tache d’huile, les cataractes d’origine métabolique peuvent régresser avec un régime d’exclusion
Plus rarement, les étiologies possibles sont :
-
déficit en glucose 6 phosphate déshydrogénase (G6PD);
-
diabète sucré car les hypoglycémies répétées peuvent entraîner une cataracte rapidement évolutive;
-
rachitisme ou autres anomalies phosphocalciques, pseudo-parathyroïdie ou syndrome d’Albright
Des cataractes ont également été décrites :
-
dans la maladie de Fabry : déficience en alpha-galactosidase avec cornea verticillata;
-
dans la mannosidérose : déficience en alpha-mannosidase avec opacités cornéennes ponctuées et cataracte.
Classiquement, la plus fréquente des infections fœtomaternelles était la rubéole (peu d’atteinte depuis la vaccination des jeunes filles) entraînant : surdité, cataracte, anomalies cardiaques et parfois retard mental; une uvéite est souvent associée contre-indiquant l’implantation.
Les autres infections sont plus rares : toxoplasmose, toxocarose, cytomégalovirus, herpès souvent à l’origine de cataractes associées à une microphtalmie et parfois des foyers rétinochoroïdiens.
Une cataracte totale ou partielle peut se voir dans les dysgénésies sévères du segment antérieur (aniridie) ou une anomalie du développement (colobomes choriorétiniens).
Les cataractes iatrogènes surviennent chez l’enfant plus grand après une radiothérapie ou une corticothérapie prolongée et sont le plus souvent sous-capsulaires postérieures. Elles peuvent aussi être consécutives à une chirurgie oculaire (vitrectomies, glaucomes congénitaux, etc.)
-
Les cataractes unilatérales sont le plus souvent malformatives, liées à des anomalies du développement oculaire (dysgénésies du segment antérieur, microphtalmie, persistance du vitré primitif, aniridie, etc.), ou idiopathiques si le bilan pédiatrique et l’examen des parents sont normaux.
-
Chez l’enfant plus grand, la principale étiologie est le traumatisme.
[1] Taylor D The Doyne Lecture. Congenital cataract : the history, the nature and the practice Eye 1998 ; 12 : 9-36
[2] Taylor D Lens Paediatric Ophthalmology : Blackwell (1997). 445-476
[3] Roche O, Beby F, Orssaud C, et al. Cataracte congénitale J Fr Ophtalmol 2006 ; 29 : 443-455
[4] Foster A Childhood blindness Eye 1988 ; 2 : S27-S36
[5] Rosenberg T, Flage T, Hansen E, et al. Visual impair ment in Nordic children. II. Aetiological factors Acta Ophthalmol (Copenh) 1992 ; 70 : 155-164
[6] Riise R, Flage T, Hansen E, et al. Visual impairment in Nordic children. IV. Sex distribution Acta Ophthalmol (Copenh) 1992 ; 70 : 605-609
[7] Blohme J, Tornqvist K Visual impairment in Swedish children. I. Register and prevalence data Acta Ophthalmol Scand 1997 ; 75 : 194-198
[8] Blohme J, Tornqvist K Visual impairment in Swedish children. II. Etiological factors Acta Ophthalmol Scand 1997 ; 75 : 199-205
[9] Blohme J, Tornqvist K Visual impairment in Swedish children. III. Diagnoses Acta Ophthalmol Scand 1997 ; 75 : 681-687
[10] Arnaud C, Baille MF, Grandjean H, et al. Visual impairment in children : prevalence, aetiology and care, 1976-85 Paediatr Perinat Epidemiol 1998 ; 12 : 228-239
[11] Rahi JS, Dezateux C Epidemiology of visual impairment in Britain Arch Dis Child 1998 ; 78 : 381-386
[12] Crofts BJ, King R, Johnson A The contribution of low birth weight to severe vision loss in a geographically defined population Br J Ophthalmol 1998 ; 82 : 9-13
[13] Eurocat EWG Surveillance of congenital anomalies 1980-1988 Brussels: Lechat MF (1991).
[14] Stoll C, Alembik Y, Dott B, Roth MP Epidemiology of congenital eye malformations in 131,760 consecutive births Ophthalmic Paediatr Genet 1992 ; 13 : 179-186
[15] Foster A, Gilbert C Epidemiology of visual impairment in children Paediatric Ophthalmology : Blackwell (1997). 3-12
[16] Souken P, Petrie A, Drew K Promotion of visual development of severely visually impaired babies : evaluation of a development based program Dev Med Child Neurol 1991 ; 33 : 320-335
C. Speeg-Schatz
C’est l’absence rare du cristallin par non-invagination de la placode optique dans la vésicule optique. Dans sa forme primaire, elle est associée à d’autres anomalies du développement des segments antérieur et postérieur. Il existe de rares aphaquies secondaires dans des microphtalmies.
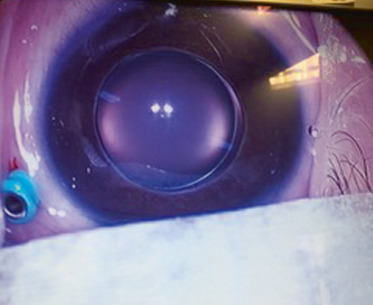
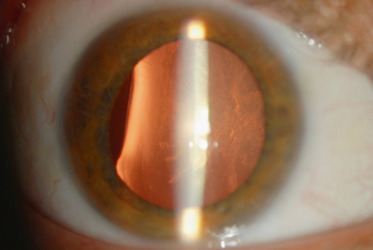
Fig. 13-14 Colobome uvéal avec encoche cristallinienne.
C’est une anomalie bilatérale du cristallin qui est de petite taille, parfois subluxé ou ectopique, au diamètre équatorial plus petit que la normale, avec étirement de la zonule visible après dilatation pupillaire (fig. 13-13). La chambre antérieure est parfois plus étroite exposant au risque de glaucome par fermeture de l’angle. Le risque est la luxation antérieure ou postérieure de ce cristallin. Cette anomalie est souvent associée à d’autres syndromes comme le Marfan, le Weill-Marchesani, l’homocystinurie, l’Alport ou le Klinefelter, etc
Cette anomalie secondaire à un colobome uvéal entraîne un aspect d’indentation de la périphérie du cristallin souvent en inféronasal (fig. 13-14 et fig. 3-34); il faut rechercher d’autres anomalies colobomateuses et une opacité cristallinienne en regard.
Les ectopies cristalliniennes (fig. 13-15) posent le problème de leur étiologie et de leur prise en charge d'abord optique, puis rééducative de l'amblyopie et chirurgicale (mode de correction de l'aphaquie) [1-5].
L'ectopie est liée à une anomalie zonulaire par distension ou rupture. Il s'ensuit un déplacement du cristallin dans un plan d'abord frontal, puis antéropostérieur. Le cristallin est clair mais peut s'opacifier secondairement.
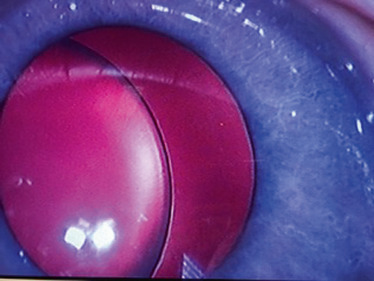
L'ectopie peut être isolée ou associée à un contexte général (Encadré 13-1) :
-
isolée : ectopie congénitale simple familiale autosomique dominante, supérieure, nasale ou temporale;
-
associée à une ectopie de la pupille : autosomique récessive, les deux ectopies se faisant dans le même axe mais en direction opposée;
-
associée à un contexte général : syndrome de Marfan, homocystinurie, syndrome d’Ehlers-Danlos, syndrome de Weil-Marchesani, déficit en sulfite oxydase.
-
Syndrome de Marfan : AD; grande taille, arachnodactylie, cyphoscoliose, anomalies cardiaques et oculaires (ectopie supérieure ++), myopie forte, baisse d’acuité visuelle. Risques : hypertonie, bascule antérieure ou postérieure du cristallin.
-
Homocystinurie : AR; déficience de la cystathionine-bêta-synthase dans le métabolisme de la méthionine (homocystine détectée dans les urines). Retard mental, troubles comportementaux, thromboses artérielles et veineuses, genu valgum, scoliose et ectopie cristallinienne (inférieure) dans 90 % des cas. Risques : hypertonie, luxation antérieure du cristallin.
-
Syndrome d’Ehlers-Danlos : AR; cutis elastica, hyperlaxité ligamentaire, sclérotiques bleues, kératocône, stries angioïdes, rares ectopies cristalliniennes.
-
Syndrome de Weil-Marchesani : AR; microsphérophaquie et brachymorphie, brachycéphalie, doigts courts et épais. Risques : hypertonie aiguë et luxation antérieure du petit cristallin
-
Déficit en sulfite oxydase : subluxation cristallinienne, rigidité musculaire progressive, rapidement léthale.
AD : autosomique dominant; AR : autosomique récessif.
[1] Nemet AY, Assia EI, Apple DJ, Barequet IS Current concepts of ocular manifestations in Marfan syndrome Surv Ophtahlmol 2006 ; 51 : 561-575
[2] Konradsen TR, Zetterström C A descriptive study of ocular characteristics in Marfan syndrome Acta Ophthalmol 2013 ; 91 : 751-755
[3] Dean J Marfan Sybdrome : clinical diagnosis and management Eur J Human Genet 2007 ; 15 : 724-733
[4] Faivre L, Collod-Beroud G, Adès J, et al. The new Ghent criteria for Marfan syndrome : what do they change? Clin Genet 2012 ; 81 : 433-442
[5] Rodrigues AC, Trivedi RH, Wilson ME Subluxation of the crystalline lens : a no-ring approach J Pediatr Ophthalmo Strabismus 2012 ; 49 : 157-163
A. Sauer
La cataracte congénitale bilatérale est la première cause de cécité chez l'enfant dans le monde (5 à 20 % des causes de cécité, soit 200 000 enfants aveugles) dans le monde. Dans les pays développés, l'incidence de la cataracte congénitale varie de 1 à 4 cas pour 10 000 naissances. Les étiologies des cataractes congénitales sont multiples et variées. Elles ont fait l'objet de nombreuses revues de la littérature. Les données de l'interrogatoire ou de l'examen clinique (ophtalmologique et pédiatrique) sont caractéristiques de certaines étiologies de cataractes congénitales et permettent un diagnostic étiologique dans plus de la moitié des cas [1-3 ].
Les causes les plus classiques sont les cataractes héréditaires sur un mode autosomique dominant, les troubles métaboliques, les cataractes chromosomiques et syndromiques, les infections intra-utérines, les traumatismes et la persistance hyperplasique du vitré primitif [1-3 ]. Les étiologies des cataractes de l'enfant sont décrites dans le Tableau 13-1.
L’hérédité sur un mode autosomique dominant est l’étiologie la plus fréquente de la cataracte congénitale. Dans ces formes, l’association à une microphtalmie est courante. Une cataracte asymptomatique doit toujours être recherchée chez les parents, car l’hétérogénéité phénotypique est une caractéristique des transmissions autosomiques dominantes. Cette hétérogénéité se manifeste aussi au niveau intra-individuel avec une variabilité de la présentation clinique entre les deux yeux. Dans le cadre d’une hérédité autosomique dominante, toutes les formes cliniques de cataractes sont décrites [2, 3].
La transmission autosomique récessive est plus rare et doit être suspectée en cas de consanguinité ou de plusieurs cas atteints avec des parents sains. Parmi les causes d’hérédité autosomique récessive, la galactosémie est une des causes notables [4].
Le syndrome de Lowe est la cause la plus classique d'hérédité liée à l'X. Les associations classiques en cas de syndrome de Lowe sont l'hypotonie, le retard mental, l'amino-acidurie, des malformations faciales (aspect joufflu et front proéminent). Le cristallin est en général de petite taille avec une réduction de son diamètre antéropostérieur. La cataracte est en général corticale ou sous-capsulaire puis s'étend progressivement au noyau. Une dysgénésie du segment antérieur est souvent notée. L'association à un glaucome est très fréquente. Le pronostic de ces cataractes est souvent très réservé même en cas de prise en charge optimale [5].
Le syndrome de Nance-Horan est une autre cause de cataracte liée à l'X (association avec un retard de développement, et des anomalies faciales comme des dents surnuméraires ou des oreilles proéminentes). La mutation génétique a été identifiée comme suit : Xp22.2-p22.3 [6].
Le syndrome de Lenz (récessif lié à l'X) peut aussi être associé à une cataracte. L'examen ophtalmologique retrouve en général une microphtalmie (colobomateuse dans 75 % des cas) et un ptosis. Au niveau général, un retard mental est souvent noté et associé à des anomalies dentaires et auriculaires, des fentes labiales ou labiopalatines, des malformations urogénitales et squelettiques [4].
Une cataracte congénitale peut se rencontrer au cours du syndrome d’Hallermann-Streiff-François. Ce syndrome est caractérisé par un visage typique (nez en bec et hypoplasie de la mandibule), une petite taille harmonieuse, une hypotrichose, une microphtalmie avec cataracte congénitale. Environ 15 % des patients ont un déficit intellectuel. Moins de 100 cas ont été décrits à ce jour dans le monde, la grande majorité des cas étant sporadique. Les bases génétiques de la maladie sont encore inconnues. Le diagnostic différentiel doit être fait avec les autres syndromes pro-géroïdes (syndrome d’Hutchinson-Gilford, syndrome de Werner, dysplasie acromandibulaire) et avec la dysplasie oculo-dento-digitale [4, 7].
Les maladies métaboliques, telles que la galactosémie (évoquée précédemment), le diabète, l’hypoglycémie et le déficit en galactokinase, sont des causes classiques de cataractes congénitales.
La galactosémie est secondaire à une mutation sur le gène codant la galactose-1-phosphate-uridyl-transférase (GALT) sur le bras court du chromosome 9. Dans près de 60 % des cas, la mutation est sur l'exon 6 (Q188R). Les enfants homozygotes pour cette mutation n'ont aucune activité GALT et présente dès la petite enfance une cataracte, des symptômes digestifs sévères (nausées, vomissements, hépatomégalie) et des septicémies à bactéries à Gram positif. Les patients hétérozygotes ont des signes digestifs plus discrets et présentent en général une cataracte à l'adolescence.
La cataracte de la galactosémie est centrale en gouttelettes d'huile, avec une extension progressive responsable d'erreurs réfractives majeures. Elle est bien mise en évidence en rétro-illumination. En l'absence de traitement, ces gouttelettes vont fusionner et mener à une cataracte lamellaire puis totale par accumulation intracristallinienne de galactitol. Un régime adapté précoce supprimant le galactose permet la guérison de la cataracte sans recours à la chirurgie, avec cependant de nombreuses erreurs réfractives résiduelles. La compliance au traitement est contrôlée par le dosage du galactose-1-phosphate sérique [4, 8, 9].
La maladie de Wilson est une affection génétique rare du métabolisme du cuivre qui s'accumule principalement dans le foie et le système nerveux central. La maladie débute chez l'enfant, l'adolescent voire l'adulte jeune. De transmission autosomique récessive, l'incidence de la maladie de Wilson est de 1/40 000 naissances en France. On estime ainsi que 1000 à 1500 personnes sont atteintes de maladie de Wilson en France. Le gène muté est –, situé sur le chromosome 13 en 13q14.3-q21.1. Plus de 200 mutations de ce gène sont décrites.
Les principaux signes cliniques sont hépatiques (hépatomégalie puis hépatite voire cirrhose) et neuropsychiatriques (tremblements et mouvements anormaux, troubles de l'élocution et de la déglutition, encéphalopathie). Au niveau ophtalmologique, l'atteinte caractéristique est l'anneau cornéen de Kayser-Fleischer (anneau verdâtre circulaire) présent chez 70 % des malades et 100 % des malades avec des manifestations neurologiques. Plus rarement, une cataracte en fleur de tournesol est retrouvée. Le traitement (chélateur du cuivre ou zinc) est d'autant plus efficace qu'il est débuté précocement [10, 11].
Les mitochondropathies peuvent aussi être à l’origine de cataracte. Elles associent en général dès la petite enfance des manifestations multisystémiques (neurologiques, cardiaques, musculaires) et des atteintes oculaires multiples et variées : rétinites pigmentaires, cataractes, cécités corticales, neuropathies optiques, ophtalmoplégies. Le diagnostic est évoqué sur l’association entre des atteintes ophtalmologiques et des atteintes neurologiques cardiaques et/ou musculaires. La confirmation génétique doit alors toujours être recherchée [4, 10].
La xanthomatose cérébrotendineuse est une maladie autosomique récessive due à un défaut de l’hydroxylase mitochondriale (codée par un gène sur le chromosome 2). Elle se manifeste par des signes neurologiques (ataxie, démence) et des xanthomes tendineux. La cataracte survient le plus souvent après l’âge de 10 ans; elle est en général bilatérale, irrégulière, corticonucléaire ou sous-capsulaire [4, 10]
Les enfants avec une hypocalcémie présentent en général des retards de développement et des troubles du comportement. La cataracte se développe en raison d’un trouble de la perméabilité capsulaire; elle est en général corticale puis devient lamellaire. La calcémie et la phosphorémie devraient être dosées chez tout enfant avec une cataracte bilatérale [4, 10].
Le diabète est rarement responsable de cataracte chez l’enfant. Ces cataractes surviennent en général à l’adolescence. À l’inverse, l’hypoglycémie néonatale (survenant préférentiellement chez les bébés avec un petit poids de naissance) peut conduire à des opacités cristalliniennes. Ces opacités sont dans la majorité des cas réversibles [4, 10].
Le syndrome cataracte-hyperferritinémie est caractérisé par une cataracte de développement précoce, bien que généralement absente à la naissance, associée à une élévation persistante du taux de ferritine sérique, en l’absence de surcharge en fer. La cataracte est généralement en « mie de pain » . La prévalence reste à déterminer précisément, mais elle est d’au moins 1 cas pour 200 000. Ce syndrome se transmet sur le mode autosomique dominant. Il est dÛ à la présence d’une mutation dans un élément de régulation traductionnelle du gène de la sous-unité L-ferritine (FTL pour ferritin light chain), situé en 19q13.4-qter. Cette mutation conduit à une élévation de la ferritine sérique, indépendante de toute surcharge en fer. Il n’existe actuellement pas de traitement mais les saignées doivent être évitées. Elles sont très mal tolérées, car les patients n’ont pas de surcharge en fer. Ce syndrome n’a pas d’autre conséquence que les cataractes et le pronostic est favorable [4, 10].
Des anomalies chromosomiques telles que les trisomies 13, 18, 21 ou les translocations (5p, 18p, 18q) sont décrites et peuvent être associées à des cataractes [3, 4]
Les enfants avec une trisomie 21 développent en général une cataracte au cours de l’enfance, même si des cataractes précoces chez les nourrissons sont parfois rencontrées. La trisomie 21 est une anomalie chromosomique définie par la présence d’un troisième exemplaire, en totalité ou en partie, du chromosome 21. La trisomie 21 n’est pas une anomalie rare (1/2000 naissances vivantes en France), mais son incidence à la naissance a diminué significativement dans plusieurs pays, après la mise en place du dépistage prénatal. Les signes classiques de trisomie 21 sont une déficience intellectuelle variable, souvent légère, une hypotonie musculaire et une laxité articulaire quasi constantes. Les particularités morphologiques (fentes palpébrales en haut et en dehors, épicanthus, nuque plate, visage rond, nez petit, pli palmaire unique bilatéral) sont évocatrices mais non pathognomoniques. Les autres principales complications incluent : malformations cardiaques (canal atrioventriculaire) et digestives (atrésie duodénale), maladie de Hirschsprung, petite taille, syndrome de West, épilepsies, leucémies, apnées du sommeil, pathologies auto-immunes et endocriniennes, vieillissement précoce et maladie d’Alzheimer. Le caryotype permet de poser le diagnostic [12].
La trisomie 18 est due à la présence d'un chromosome 18 supplémentaire. L'incidence varie de 1/6000 à 1/8000 naissances. Plus de 95 % des fœtus atteints décèdent in utero. Le pronostic des enfants naissants avec une trisomie 18 est très péjoratif. Dès les premières semaines de vie, une hypotonie majeure s'installe. Un faciès caractéristique est observé (dolichocéphalie, micro-rétrognathie, hypertélorisme, oreilles mal ourlées et anguleuses dites faunesques). Les pieds sont en varus équin; les doigts de la main se chevauchent. Les malformations sont nombreuses et souvent sévères : oculaires (microphtalmie, colobome, cataracte), cardiaques (quasi constantes), digestives (atrésie œsophagienne, malformation anorectale), rénales (hydronéphrose, agénésie uniou bilatérale du rein).
La trisomie 18 peut être suspectée en cours de grossesse à l'échographie (retard de croissance, malformations, kystes multiples des plexus choroïdes, etc.) et confirmée par le caryotype fœtal. La prise en charge médicale de la trisomie 18 est limitée aux soins de support et de confort. La trisomie 18 est très sévère : 90 % des enfants décèdent avant 1 an de complications cardiaques, rénales, neurologiques ou de surinfections [4].
La trisomie 13 est une anomalie chromosomique due à la présence d'un chromosome 13 supplémentaire. Elle est caractérisée par l'association de malformations cérébrales très sévères (holoprosencéphalie), de dysmorphie faciale (hypotélorisme, agénésie prémaxillaire, voire cébocéphalie ou cyclopie), d'anomalies oculaires (colobome, anophtalmie), de polydactylie postaxiale, de malformations viscérales (cardiopathie) et d'un retard psychomoteur très sévère. Son incidence est estimée entre 1/8000 et 1/15 000 naissances. Plus de 95 % des fœtus atteints décèdent in utero.
La trisomie 13 peut être suspectée en cours de grossesse à l'échographie (holoprosencéphalie, polydactylie) et confirmée par le caryotype fœtal. La prise en charge médicale de la trisomie 13 est limitée aux soins de support et de confort. La trisomie 13 est très sévère; la moitié des enfants décèdent le premier mois et 90 % avant 1 an de complications cardiaques, rénales ou neurologiques [4].
Des cataractes ont été décrites dans certains cas de translocations chromosomiques. La plus connue est la translocation 5p- ou syndrome du cri du chat. Les caractéristiques cliniques comprennent un cri monochromatique aigu, une microcéphalie, une arête nasale large, un épicanthus, une micrognathie, des anomalies des dermatoglyphes ainsi qu'un retard mental et psychomoteur important. L'incidence à la naissance est comprise entre 1/15 000 et 1/50 000 enfants nés vivants.
La monosomie 18p est une anomalie chromosomique due à une délétion totale ou partielle du bras court du chromosome 18. L'incidence est d'environ 1/50 000 naissances viables. Le syndrome dysmorphique est en général discret. Les signes cliniques principaux sont une petite taille, un visage rond avec un philtrum court, une ptose palpébrale et de grandes oreilles décollées. Le retard mental est modéré. Dix à 15 % des patients sont atteints de graves malformations du cerveau et du visage de type holoprosencéphalie [4].
Une cataracte congénitale ou infantile peut être découverte dans toute une série de syndromes avec retard mental, dont quelques-uns sont évoqués dans les lignes ci-dessous [3, 4] :
-
syndrome de Martsolf (20 cas décrits) qui comporte un retard mental, un hypogonadisme hypogonadotrope et aussi des malformations faciales (micrognathie, brachycéphalie, maxillaire plat, fentes labiopalatines);
-
syndrome de Marinesco-Sjögren (moins de 1 naissance sur 100 0000) associant cataracte et ataxie cérebelleuse;
-
chondrodysplasie ponctuée où des malformations faciales et des membres sont retrouvées;
-
syndrome cérébro-oculo-facio-squelettique ou COFS (environ 10 cas décrits) qui regroupe dysmorphie faciale (microcéphalie congénitale avec microphtalmie, suture métopique proéminente, micrognathisme), arthrogrypose, retard de croissance staturo-pondéral, hypotonie axiale et hypertonie périphérique, photosensibilité cutanée, surdité de perception et rétinopathie pigmentaire.;
-
syndrome de Czeizel-Lowry où une maladie de Legg-Calve-Perthes, ou ostéochondrite primitive de hanche, est toujours associée;
-
des cataractes congénitales ont été également rapportées de manière sporadique dans les syndromes de Bardet-Biedl, Cockayne, Hallgren, Sphrintzen, Scwartz-Jampel, etc.
Une infection maternofœtale doit être évoquée en cas de cataracte dense uni- ou bilatérale centrale. L’interrogatoire s’efforcera de rechercher un syndrome grippal et une éruption cutanée lors de la grossesse. Un bilan sérologique chez la mère et le nouveau-né, à la recherche d’une infection par la rubéole, la toxoplasmose, l’herpès, les oreillons, la syphilis ou le cytomégalovirus est indiqué. De plus, en cas de suspicion de maladie infectieuse, l’analyse virologique et parasitologique de l’humeur aqueuse peut être réalisée, avec une sensibilité cependant très limitée. L’imputabilité des causes infectieuses est souvent bien difficile à confirmer en dehors des phénotypes extrêmes, où le pronostic visuel est très réservé en raison des atteintes associées (rétiniennes et neurologiques notamment) [2-4].
La persistance hyperplasique du vitré primitif (PHVP) est une entité définie par une prolifération rétrocristallinienne sur un œil microphtalme avec une néovascularisation irienne, une chambre antérieure étroite, une élongation des procès ciliaires. La PHPV est unilatérale dans plus de 90 % des cas. Un strabisme et un nystagmus sont fréquemment associés. Le pronostic fonctionnel de la PHVP est relativement mauvais en raison d’une amblyopie profonde au moment du diagnostic et de l’association fréquente à des complications de type glaucome ou décollement de rétine [13]
Les cataractes traumatiques chez l'enfant sont essentiellement liées à des blessures par projectile. Une plaie du globe est associée dans 1 cas sur 4. La prise en charge de ces traumatismes est sensiblement la même que chez l'adulte, avec les difficultés liées à la fragilité zonulaire ou à une effraction capsulaire. Les principales différences résident dans le choix de l'implant et le suivi ultérieur des risques de glaucome secondaire et de décollement de rétine, dont les incidences semblent beaucoup plus importantes chez les enfants [14].
Enfin, les inflammations intra-oculaires (par elles-mêmes ou par corticotoxicité) sont aussi responsables de cataractes infantiles [4].
Le bilan étiologique devant une cataracte congénitale est présenté dans Encadré 13-2.
Échographie cardiaque.
Bilan sanguin :
dosage de l’activité de la galactokinase ;
bilan sérologique infectieux si suspicion spécifique en cas de signes oculaires ou systémiques associées (TORSCH = toxoplasmose- oreillons-rubéole-syphilis-cytomégalovirus-herpès) ;
glycémie.
Dosage urinaire des acides aminés.
Le bilan clinique et paraclinique mené chez un enfant présentant une cataracte (hors cataracte traumatique, dont le bilan est le même que celui de l’adulte) est le suivant.
Un interrogatoire précis est réalisé à la recherche d’un syndrome infectieux, d’une exposition à un toxique ou à des rayonnements ionisants pendant la grossesse. Un examen (antécédents et biomicroscopie) des parents est indiqué afin de déterminer la présence d’une cataracte.
Des signes associés de glaucome (augmentation de la pression intra-oculaire, mégalocornée, œdème de cornée) ou de dysgénésies du segment antérieur sont à rechercher. L’examen du fond d’œil peut permettre de mettre en évidence des lésions associées, comme une persistance hyperplasique du vitré primitif, une inflammation vitréenne voire un rétinoblastome. À défaut, une échographie en mode B est toujours indiquée. Des examens électrophysiologiques (électrorétinogramme ou potentiels évoqués visuels) sont parfois utiles pour éliminer une autre cause de cécité.
Un examen systémique par un pédiatre doit toujours être réalisé afin de rechercher des malformations associées et éventuellement orienter vers une cataracte syndromique. Ce bilan est indispensable dans les cataractes bilatérales et les ectopies cristalliniennes. Un bilan clinique et échographique par un cardiopédiatre est recommandé pour dépister des malformations cardiaques (syndrome de Sengers par exemple associant une myocardiopathie à une cataracte bilatérale) et vérifier l’absence de contre-indication à une anesthésie générale.
Des examens sanguins sont parfois réalisés. Dans certains pays (notamment les États-Unis, l’Angleterre ou l’Allemagne), un dosage de l’activité de la galactokinase est systématique.
Par ailleurs, de nombreuses équipes pratiquent encore un bilan sérologique de type TORSCH (toxoplasmose-oreillons-rubéole-syphilis-cytomégalovirus-herpès). Cette pratique est discutable car peu rentable (les micro-organismes représentent moins de 1 % des cataractes congénitales) et l’imputabilité du micro-organisme dans l’apparition de la cataracte est souvent douteuse. Ce bilan sérologique devrait être réservé aux cas avec microphtalmie ou atteintes rétiniennes associées évocatrices.
En cas d’anomalies de l’examen systémique pédiatrique, on peut demander une calcémie et une phosphorémie (hypocalcémie, hypoparathyroïdie), une glycémie (diabetes mellitus), un dosage urinaire de certains acides aminés (syndromes de Lowe ou d’Alport). Ces examens sont inutiles chez un enfant en bonne santé présentant une cataracte unilatérale [4].
Les causes de cataractes congénitales et infantiles sont nombreuses et variées. Un examen ophtalmologique et pédiatrique complet permet d’orienter le bilan étiologique.
[1] Foster A, Gilbert C, Rahi J Epidemiology of cataract in childhood : a global perspective J Cataract Refract Surg 1997 ; 23 : 601-604
[2] Chan WH, Biswas S, Ashworth JL, Lloyd IC Congenital and infantile cataract : aetiology and management Eur J Pediatr 2012 ; 171 : 625-630
[3] Wirth MG, Russell-Eggitt IM, Craig JE, et al. Aetiology of congenital and paediatric cataract in an Australian population Br J Ophthalmol 2002 ; 86 : 782-786
[4] Hoyt C, Taylor D Pediatric ophthalmology and strabismus : Elsevier Saunders (2013).
[5] Loi M Lowe syndrome Orphanet J Rare Dis 2006 ; 1 : 16
[6] Toutain A, Ayrault AD, Moraine C Mental retardation in Nance-Horan syndrome : clinical and neuropsychological assessment in four families Am J Med Genet 1997 ; 71 : 305-314
[7] Nicholson AD, Menon S Hallerman-Streiff syndrome J Postgrad Med 1995 ; 41 : 22-23
[8] Endres W, Shin YS Cataract and metabolic disease J Inherit Metab Dis 1990 ; 13 : 509-516
[9] Karadag N, Zenciroglu A, Eminoglu FT, et al. Literature review and outcome of classic galactosemia diagnosed in the neonatal period Clin Lab 2013 ; 59 : 1139-1146
[10] Negahban K, Chern K Cataracts associated with systemic disorders and syndromes Curr Opin Ophthalmol 2002 ; 13 : 419-422
[11] Ram J, Gupta A Kayser-Fleischer ring and sunflower cataract in Wilson disease JAMA Ophthalmol 2014 ; 132 : 873
[12] Creavin AL, Brown RD Ophthalmic abnormalities in children with Down syndrome J Pediatr Ophthalmol Strabismus 2009 ; 46 : 76-82
[13] Skalicky SE, White AJ, Grigg JR, et al. Microphthalmia, anophthalmia, and coloboma and associated ocular and systemic features: understanding the spectrum JAMA Ophthalmol 2013 ; 131 : 1517-1524
[14] Khokhar S, Gupta S, Yogi R, et al. Epidemiology and intermediate-term outcomes of open- and closed-globe injuries in traumatic childhood cataract Eur J Ophthalmol 2014 ; 24 : 124-130
D. Thouvenin, C. Speeg-Schatz
Opérer une cataracte chez un enfant ne consiste pas à simplement appliquer une technique chirurgicale adaptée [1-4]. En premier lieu, cela s'intègre dans une stratégie thérapeutique globale. Ensuite, l'acte chirurgical, qui a bénéficié des progrès de la chirurgie du segment antérieur de l'adulte, est spécifique à l'enfant. Chaque étape de sa réalisation peut varier en fonction du cas. Prendre en charge ce type de chirurgie va donc bien au-delà d'une simple maîtrise de la chirurgie du segment antérieur, ce qui signifie :
-
connaître les étapes du développement visuel de l’enfant et les conséquences potentielles d’une privation visuelle à tout stade du développement;
-
être capable de poser l’indication au moment opportun selon le cas, ce qui est sensiblement différent de chez l’adulte;
-
s’adapter aux spécificités anatomiques de l’œil de l’enfant;
-
être prêt à s’adapter rapidement, plus fréquemment que chez l’adulte, à des complications peropératoires ou des particularités découvertes durant l’intervention ou à des situations rendant la chirurgie plus complexe : uvéite, traumatisme, suites de décollement de rétine;
-
être rapide et précis. Il est montré que plus le geste est rapide et atraumatique, plus les suites opératoires seront simples. Une courbe d’apprentissage existe. La chirurgie des cataractes de l’enfant est réservée à des chirurgiens expérimentés et la chirurgie des plus jeunes (moins de 6 ans) devrait être laissée à des chirurgiens spécialistes;
-
intégrer l’acte chirurgical dans une stratégie globale de traitement, dont le contexte, le type de cataracte, le choix du traitement de l’aphaquie, le traitement de l’amblyopie, la connaissance des suites possibles et des complications et de leur gestion,
-
s’impliquer à long terme dans le suivi de l’enfant : le plus souvent, c’est le chirurgien qui sera le référent de la suite du traitement pour l’enfant, sa famille et l’équipe médicale en charge.
Si la chirurgie de cataracte d'un grand enfant peut être envisagée par un chirurgien habitué au segment antérieur, l'intervention d'une cataracte chez un jeune enfant doit être confiée à une équipe ophtalmopédiatrique aguerrie. Elle assumera un suivi prolongé, en coordination éventuelle avec une équipe ophtalmo-orthoptique s'occupant plus étroitement de l'enfant, proche du domicile.
Une cataracte infantile ne s'opère pas toujours dès son diagnostic. La cataracte obturante totale du nouveau-né s'oppose pour cela aux cataractes partielles qui peuvent rester stables toute la vie (cataractes pyramidales antérieures par exemple) ou évoluer progressivement (cataractes capsulaires ou sous-capsulaires postérieures ou traumatiques par exemple). Le chirurgien ophtalmopédiatre doit donc toujours opposer la nécessité de rétablir des milieux optiquement utilisables avec les conséquences irréversibles de l'intervention (perte de l'accommodation, évolution réfractive, etc.) et ses risques.
Les cataractes congénitales totales uni- ou bilatérales doivent être opérées rapidement, car les premières semaines de vie sont cruciales pour les acquisitions visuelles. En effet, une cataracte bilatérale totale dès la naissance provoque le plus souvent l'apparition d'un nystagmus pendulaire et une cataracte congénitale unilatérale totale est génératrice d'amblyopie profonde. La précocité de l'intervention influe sur la qualité de l'évolution visuelle ultérieure. Toutefois, il est aussi montré qu'une intervention très précoce, durant le premier mois de vie, est d'une part, beaucoup plus complexe en raison de tissus immatures et d'autre part, pourvoyeuse de complications potentiellement cécitantes beaucoup plus fréquentes notamment de glaucomes de l'aphaque et du pseudo-phaque. Le meilleur moment pour opérer ces cataractes congénitales totales précoces semble finalement se placer entre 6 et 8 semaines de vie. Les cataractes bilatérales incomplètes avec une périphérie du cristallin clair ou une visibilité même partielle du fond d'œil (FO) gagneront même à être opérées après 3 mois d'âge si elles sont symétriques. Une dilatation pupillaire médicamenteuse jusqu'à l'intervention permet une stimulation lumineuse rétinienne sans doute très utile en attendant l'intervention.
Dans le cas des cataractes bilatérales, l'indication chirurgicale est indiscutable dès qu'elles sont obturantes (FO mal analysé en ophtalmoscopie indirecte) ou qu'elles provoquent une gêne visuelle importante malgré une correction optique adéquate. Cette gêne visuelle varie selon l'âge : gêne visuelle empêchant des acquisitions ou activités normales chez un petit, baisse d'acuité visuelle limitant les possibilités scolaires et sociales chez un plus grand, photophobie invalidante. Une asymétrie franche entre les deux yeux peut conduire à avancer l'intervention sur l'œil le plus atteint pour éviter une amblyopie, toujours après essai de traitement médical par occlusion. La question d'une intervention bilatérale d'emblée se pose en cas de risque anesthésique important ou dans les régions et pays où l'accès aux soins est complexe. Si les précautions d'usage sont suivies, le taux de complication n'est pas plus important qu'en cas de chirurgie unilatérale, mais ce n'est pas un choix habituel si l'accès au soin est simple. En revanche, le délai entre l'intervention de chaque œil sera d'autant plus court que l'enfant est jeune, afin de ne pas provoquer une amblyopie unilatérale.
Pour les cataractes unilatérales, la décision est parfois plus complexe. L'indication est évidente en cas de cataracte obturante, après avoir vérifié l'intégrité du segment postérieur par une échographie. En revanche, quand la cataracte n'est pas obturante, il faut savoir faire la part des choses entre la baisse visuelle réellement liée à la perte de transparence (part organique) et celle liée à l'amblyopie fonctionnelle. Dans ces cas, on peut approcher le caractère réellement obturant de l'opacité en se demandant ce qu'un adulte verrait au travers ou en tentant d'analyser les détails du FO en ophtalmoscopie indirecte, mais cela reste très subjectif. Le mieux est de réaliser un test thérapeutique d'occlusion totale jusqu'à récupération d'une fixation stable chez l'enfant préverbal ou d'une acuité visuelle maximale, sans souffrance de l'œil sain. Si le résultat est satisfaisant, on poursuit simplement le traitement d'amblyopie jusqu'en fin de période sensible. Si le résultat est insuffisant ou instable, il vaut mieux envisager l'intervention en cours de rééducation. Dans tous les cas, les parents doivent être prévenus que l'intervention ne servira à rien sans le traitement prolongé de l'amblyopie dont on sait maintenant que les résultats sont loin d'être négligeables, même en cas de cataracte congénitale précoce obturante.
L’opération d’une cataracte chez un enfant est un événement familial qui dépasse largement celui d’une opération d’une cataracte chez l’adulte. C’est un des rôles du chirurgien ophtalmopédiatre de savoir gérer avec clarté la préparation de l’enfant et de la famille. La place des explications est fondamentale. Celles-ci ne se limitent pas à la technique chirurgicale et aux risques directs mais s’étendent à l’ensemble de la stratégie thérapeutique. On aborde donc :
-
l’intervention et ses risques;
-
le choix du moment de l’intervention;
-
les contraintes de surveillance médicale et visuelle pendant les années à venir;
-
le choix du mode de correction de l’aphaquie (lunettes, lentilles, implant intra-oculaire);
-
le traitement de l’amblyopie;
-
éventuellement, l’évaluation du pronostic visuel futur en s’appuyant sur les statistiques rapportées au cas de l’enfant.
Cet examen est le plus souvent suffisant pour décider ou non l'intervention et apprécier la complexité ou les spécificités du cas. Il sera de toute façon complété sous anesthésie.
-
Examen visuel classique :
-
comportement visuel, photophobie, appréciation du retentissement visuel général de la cataracte;
-
monoculaire, selon l’âge, avec la présence ou non d’une amblyopie;
-
binoculaire, présence d’un strabisme ou non et son type. État de la vision binoculaire en l’absence de strabisme;
-
présence d’un nystagmus, souvent pendulaire dans les cataractes bilatérales précoces ou manifeste latent en cas de cataracte unilatérale précoce avec éventuellement syndrome du monophtalme congénital.
-
-
Réfraction : impossible à évaluer en cas de cataracte totale mais pas en cas d’opacité partielle. Toutefois, si la cataracte s’est opacifiée progressivement, l’historique de la réfraction est intéressant à connaître.
-
Examen ophtalmopédiatrique simple :
-
contexte général de la cataracte (antécédents familiaux, pathologies associées notamment);
-
évaluation simple du segment antérieur à la lampe à fente quand c’est possible, à l’ophtalmoscope toujours;
-
FO en ophtalmoscopie indirecte. On peut ne rien voir en cas de cataracte obturante ou voir simplement la périphérie en cas de cataracte zonulaire ou nucléaire ou encore voir le pôle postérieur plus ou moins flouté et parfois même une simple ombre portée de la cataracte sous-capsulaire postérieure. Si le FO est très bien analysé, il faut remettre en question l’indication de la chirurgie. Une cataracte minime associée à une très mauvaise acuité visuelle est souvent synonyme d’amblyopie profonde. Si l’âge le permet, un traitement d’amblyopie doit donc être réalisé avant intervention.
-
-
Échographie en mode B si le FO est invisible. Une échographie haute fréquence peut être utile en cas de dysgénésie du segment antérieur associée.
-
Biométrie si possible, avec kératométrie et longueur axiale au minimum.
-
La réalisation d’examens électrophysiologiques est intéressante mais ne remet pas en question l’indication opératoire. Elle a plus un intérêt pronostique.
En général préopératoire immédiat, parfois préalable si des compléments diagnostiques sont indispensables à la discussion de l’intervention (notamment quant à un doute sur l’état anatomique ou fonctionnel du segment postérieur). On réalise :
-
la prise du tonus oculaire;
-
un examen au microscope du segment antérieur et on complète l’examen du FO;
-
une échographie diagnostique en mode B;
-
la biométrie complète avec diamètre cornéen, kératométrie, échographie en mode A;
-
une électrophysiologie avec électrorétinogramme (ERG) et potentiels évoqués visuels (PEV) si nécessaire.
La technique de référence actuelle est celle de phaco-aspiration par voie antérieure avec capsulotomie postérieure et vitrectomie antérieure. Elle permet, en outre, d’implanter d’emblée. Elle bénéficie de tous les progrès de la microchirurgie du segment antérieur [5-7]. Dans certains cas, un abord par la pars plana peut être utile et les variantes de la chirurgie dépendent alors beaucoup des habitudes de chaque chirurgien. L’abord postérieur bénéficie bien sÛr des progrès technologiques avec l’apport des voies d’abord 25 G et de l’endoscopie qui peut être utile dans certains cas. Quelle que soit la voie d’abord, il est important de laisser une couronne périphérique de capsule antérieure et postérieure pour l’implantation qu’elle soit initiale en intercapsulaire ou secondaire dans le sulcus.
L’incision doit être adaptée à l’œil d’enfant : cornée épaisse et molle, chambre antérieure étroite, accès difficile en cas de myosis, astigmatisme inverse fréquent. Elle doit « aider » la chirurgie pour éviter le prolapsus irien; ses dimensions sont adaptées aux instruments d’aspiration. Elle peut être sclérale antérieure, moins génératrice de taies cornéennes « iatrogènes » mais la tendance est aux incisions cornéennes, de taille et de construction similaire à l’adulte et adaptée aux micro-instruments et aux dispositifs d’implantation. La réduction de leur taille (1,5 à 2,3 mm) les rend moins astigmatogènes et permet de les positionner à 2 h et 10 h. Elles seront systématiquement refermées en raison de l’élasticité de la cornée qui compromet l’étanchéité, surtout chez des enfants qui se frottent les yeux.
Le matériau viscoélastique est fondamental et son utilisation a bien sÛr bouleversé la technique chirurgicale. Il doit avoir une haute viscosité et une forte cohérence. Il aide au maintien et à l’élargissement de la mydriase. Il aide au traitement des capsules, refoule le vitré (pression élevée, mollesse sclérale) et ouvre les feuillets capsulaires pour l’implantation. En l’absence de dilatation pupillaire suffisante, celle-ci sera obtenue par des moyens mécaniques, crochets ou autres, afin de ne pas compromettre la qualité de l’intervention.
La réalisation d’un capsulorhexis circulaire continu (CCC) conditionne la suite de l’intervention, notamment l’implantation. Elle est délicate car la capsule est très élastique et sous tension et la tendance est au refend radiaire. De plus, la mydriase est souvent très relative. Enfin, dès que la visibilité de la capsule n’est pas parfaite, il faut s’aider du bleu trypan pour la réalisation du CCC. Il est plus simple dans un premier temps de le réaliser à la pince, mais on peut le réaliser à l’aiguille avec un peu d’habitude. On réalise une ponction au cystotome à 6 h, puis le CCC est réalisé par tractions radiaires et légèrement rétrogrades, en rattrapant fréquemment le bord du capsulorhexis, car le refend a facilement tendance à filer en périphérie. Un diamètre de 6 mm est satisfaisant car la capsule ira se souder autour de l’éventuel implant sur la capsule postérieure. Il existe de nombreuses variantes de cette ouverture : technique pushpull de Nischal, en pointillé, au ciseau puis pince, au vitréotome (vitréorhexis), à la diathermie bipolaire haute fréquence et au laser femtoseconde. Chaque technique a ses avantages et ses inconvénients propres et sa courbe d’apprentissage
Même en l’absence de noyau, c’est une étape non négligeable car elle aide à bien détacher le cortex périphérique et, en aidant un bon nettoyage, limite les risques de prolifération secondaire. On l’évitera en cas de doute sur un défect de la capsule postérieure (lenticônes ou autres cataractes sous-capsulaires postérieures).
Elle ne nécessite pas d’ultrasons puisque le noyau est mou. La technique bimanuelle prend ici tout son intérêt. Il faut réaliser un pelage capsulaire très soigneux, en raison du caractère prolifératif majeur des cellules équatoriales. Il faut rester toujours prudent devant la capsule postérieure, que l’on ouvrira de toute façon mais de manière contrôlée. Les pompes à effet Venturi présentent un avantage dans certains cas complexes.
Parks a bien montré l'importance de la réaliser de manière systématique lors de la première intervention [8]. L'expérience montre que l'opacification de la capsule postérieure (OCP) est systématique jusqu'à 7 ans (sans doute plus) en raison du potentiel prolifératif des cellules souches équatoriales. Avant d'être en âge de bénéficier du laser YAG (), il faut donc préventivement l'ouvrir chirurgicalement par un CCC postérieur pour les mêmes raisons que pour l'antérieur (contention de l'implant notamment). Il faut aussi retirer le vitré antérieur et la hyaloïde qui, sinon, servent de support mécanique à une prolifération des masses équatoriales. La vitrectomie antérieure est donc systématique. Ce geste, bien contrôlé est peut-être même moins agressif qu'une capsulotomie au laser YAG. La soudure des deux capsules évite la diffusion des cellules mais elle n'est pas contrôlable. La luxation de l'optique de l'implant en arrière du CCC postérieur y aiderait, mais assez inconstamment. C'est ce qui a conduit Tassignon [9] à proposer de mettre des implants où les deux feuillets capsulaires sont enchâssés dans une rainure autour de l'optique de l'implant avec des résultats intéressants mais une courbe d'apprentissage de la technique non négligeable.
La capsule postérieure est très fine et fibrillaire. Le capsulorhexis est réalisé après ponction et injection de viscoélastique en arrière pour décoller la hyaloïde antérieure. On peut réaliser un capsulorhexis à la pince ou un vitréorhexis, en tout cas continu et circulaire. Nous le réalisons préférentiellement avant implantation. Certains chirurgiens préfèrent implanter d'abord dans le sac puis luxer l'optique et le réaliser ensuite, en arrière de l'implant.
En présence d'une persistance de la vascularisation fœtale (PVF), l'ouverture capsulaire postérieure est accompagnée d'une endocoagulation du pédicule vasculaire puis de sa section. Puis, on découpe la galette opaque de la capsule postérieure et, en fonction de sa taille et du degré de rétraction du corps ciliaire, une implantation pourra être discutée.
Après implantation ou non, il faut évacuer soigneusement le viscoélastique et remettre en myosis, par injection de myotique intracamérulaire, ce qui permet de vérifier l'absence de bride vitréenne tendue vers la kératotomie. Une iridectomie périphérique n'est pas systématique, car elle a ses propres risques et inconvénients. On la réalisera d'autant plus que l'intervention a été compliquée, que l'enfant est jeune et que le contexte est inflammatoire.
La fermeture de la kératotomie est systématique chez l'enfant, pour en augmenter l'étanchéité, la solidité et limiter le risque d'astigmatisme inverse. L'utilisation de monofilament de Nylon nécessite de rendormir l'enfant pour l'ablation des points. Nous utilisons préférentiellement le Vicryl 10/0 pour éviter cet ennui. Les points doivent être bien tendus. L'astigmatisme induit par les sutures a de toute façon tendance à s'inverser par la suite, de manière beaucoup plus fréquente que chez l'adulte.
La phase postopératoire comporte une injection sous-conjonctivale de dexaméthasone en fin d'intervention, puis à la demande en cas d'inflammation. D'autres utilisent de la triamcinolone.
La nécessité d'une injection d'antibiotique intracamérulaire n'est pas validée chez l'enfant; elle se fait hors autorisation de mise sur le marché (AMM). Certains utilisent la moxifloxacine ou encore la céfuroxime.
Des collyres antibiotiques corticoïdes et une dilatation par atropine sont prescrits pour 1 à 2 mois. La corticothérapie générale n'est pas couramment utilisée sauf intervention compliquée.
Nous recommandons un isolement relatif de l'enfant 8 jours avant et 10 jours après l'intervention afin de limiter le risque de maladie infectieuse intercurrente pendant le premier stade de la période de cicatrisation.
Une surveillance simple mais hebdomadaire de l'état local nous paraît justifiée durant le premier mois (vérification de la lueur pupillaire, de l'aspect pupillaire notamment).
Si nécessaire, l'occlusion peut être débutée dès que la transparence des milieux est suffisante.
Une correction optique provisoire peut être prescrite. Elle sera adaptée à la réfraction au moins 1 mois après intervention. L'équipement de l'aphaquie est décrit ci-dessous.
La situation se présente essentiellement dans les maladies de Marfan et dans les ectopies idiopathiques (30 % ) parfois héréditaires ou de causes variées (syndromes de Weill-Marchesani, homocystinurie, aniridie, etc.) [10]. Parfois, chez l'enfant plus grand, il peut s'agir de rares luxations traumatiques.
L'indication se pose en cas d'ectopie évolutive avec retentissement visuel non accessible à une correction optique, et nécessairement si luxation.
Le plus souvent, la zonule est assez solide pour permettre une phaco-aspiration par un petit capsulorhexis, puis le sac est retiré dans son ensemble à la pince.
L'implantation est soigneusement discutée sur ces yeux fragiles. Le sac ne peut raisonnablement pas servir de support en raison de la fragilité zonulaire. L'implantation sans support capsulaire aggrave cette fragilité oculaire. Dans la maladie de Marfan, la raison pousse à laisser l'œil non implanté en raison du fort risque de complications spontanées (glaucome, décollement de rétine, etc.). Dans les autres situations, si une implantation est discutée, il faut toujours la mettre en balance avec l'extrême fragilité de ces yeux (voir plus loin) [11, 12].
L’ablation du cristallin provoque l’aphaquie optique, la perte de l’accommodation, la perte d’un filtre protecteur pour la rétine, mais aussi, sur le plan anatomique, la perte d’une barrière entre les segments antérieur et postérieur et enfin la perte d’une certaine rigidité du globe. Chacun de ces points a ses conséquences.
Plusieurs solutions sont possibles dont aucune n’est pleinement satisfaisante et les attitudes évoluent avec le temps :
-
l’implantation intra-oculaire n’est proposée chez l’enfant que si les risques sont minimisés par une intervention simple et un état anatomique local correct. Elle devient progressivement le choix initial chez l’enfant de plus de 3 ans, mais divise les chirurgiens en dessous. En dessous de 3 ans et surtout de 1 an, une implantation traumatique peut avoir des conséquences dramatiques. Les chirurgiens anglo-saxons [13] sont beaucoup plus réservés qu’en Europe continentale, en Asie et en Inde où les résultats semblent très encourageants. L’étude nord-américaine IATS semble montrer l’absence de bénéfice visuel et le plus fort taux de complications de l’implantation chez le jeune enfant, mais la méthodologie des études est critiquable. Il semble au contraire qu’entre des mains entraînées, elle ne favorise pas les complications cécitantes et présente des avantages importants dans la gestion des enfants opérés dans les pays où l’accès aux soins est complexe [14]. L’implantation chez le jeune enfant doit être réservée à des chirurgiens habitués. La technique est décrite plus bas;
-
le port de lentilles de contact est décrit dans le chapitre 5-12. Dans le cadre de l’aphaquie, il présente de nombreux avantages dont la moindre agressivité et l’adaptabilité aux modifications réfractives. D’un autre côté, cet équipement nécessite la présence d’un ophtalmologiste habitué à ce type d’équipement et disponible, ce qui est assez rare. Par ailleurs, les parents sont confrontés au coÛt des lentilles avec pertes fréquentes et interruptions répétées des traitements d’amblyopie. On peut dire que cette solution est idéale quand les conditions sont optimales, ce qui est rare;
-
les lunettes d’aphaquie sont le moyen le plus simple de corriger l’aphaquie mais nécessite de fortes puissances des verres. C’est un moyen encore fréquemment utilisé dans les cataractes bilatérales opérées précocement. Dans les cataractes unilatérales, cela peut être un moyen utilisé transitoirement en cas de perte de la lentille pour ne pas interrompre la rééducation. L’enfant peut ne porter cette correction que lors du port de l’occlusion, même si l’habituation aux lunettes tout le temps peut être préférable. Il n’est de toute façon pas gêné par l’aniséiconie puisque dans ce type de cataracte unilatérale précoce, la vision binoculaire n’existe pas;
-
l’équipement optique complémentaire : la réfraction doit être réévaluée fréquemment car elle évolue rapidement d’autant plus que l’enfant est jeune. En cas de port de lentilles, il faut simplement adapter leurs puissances. En cas d’implantation, une hypermétropie résiduelle est volontairement laissée en place pour limiter le risque d’une myopisation majeure. Il faut en suivre l’évolution par des skiascopies répétées
La perte d’accommodation présente un des obstacles à une bonne récupération visuelle. C’est aussi un des arguments pour retarder les interventions en cas de cataracte partielle :
-
avant l’âge de 2 ans, on met en général en place une correction monofocale permettant une bonne exploration du monde proche, intérêt principal des petits enfants. Une addition de 2 ou 3 D sur la valeur de la réfraction est le choix habituel;
-
à partir de 2 ans, ou immédiatement pour certains, une addition de près est nécessaire, avec TELEX® ou montage Franklin, ou encore verres bifocaux plus tard. Les verres progressifs sont bien utilisés par les enfants mais doivent être montés assez haut pour une utilisation spontanée simple. Ils peuvent être utilisés selon les auteurs dès 2 à 6 ans. Les bifocaux sont utilisés préférentiellement dans les cas de malvoyance avec nystagmus important et avec un grand foyer inférieur;
-
les implants multifocaux sont déconseillés durant toute la période de croissance du globe et tant qu’une réfraction subjective de qualité ne peut être réalisée. Il faut les éviter en cas de myopie axiale préexistante à l’intervention, car on ne sait alors le potentiel évolutif de cette myopie. En pratique, leur utilisation peut être discutée au cas par cas après l’âge de 8 ans. Si l’indication est bien choisie, ils sont en général très bien tolérés et utilisés par les grands enfants, que ce soit en cas de cataracte uni- ou bilatérale;
-
en cas de strabisme associé à une cataracte bilatérale, l’utilisation de pénalisations alternées est une bonne solution pour permettre d’entretenir l’alternance de fixation et de proposer dans le même temps une correction pour vision de loin et de près. On alterne le port de lunettes comprenant un verre corrigé aux valeurs de la réfraction exacte et l’autre sur-corrigé de 3 D. L’inverse est réalisé sur une autre paire de lunettes et l’enfant alterne quotidiennement le port des deux lunettes.
Le cristallin filtre certaines longueurs d’onde de la lumière notamment les ultraviolets (UV). Il est probable que la perte de ce filtre puisse avoir des conséquences rétiniennes à long terme. En l’absence d’implantation ou de lentilles filtrantes, il est recommandé d’inclure dans les verres un filtre UV. L’utilité des autres filtres est discutée au cas par cas. Le filtre des bleus est souvent inclus dans les implants mais il semble diminuer la biocompatibilité des implants chez le très jeune enfant (en dessous de 3 ans).
L’aphaquie retire la barrière physique entre segment antérieur et postérieur. On n’en connaît pas bien les conséquences à long terme. La perte précoce du support mécanique zonule/cristallin a sans doute un rôle dans la myopisation parfois excessive des aphaques. Mais sa responsabilité est surtout invoquée dans la genèse des glaucomes de l’aphaque et du pseudo-phaque qui surviennent dans près de 30 % des cas opérés avant 1 an et sans doute plus, si le délai de surveillance est prolongé. Cette perte précoce modifie sans doute la croissance et l’anatomie de l’angle iridocornéen. On devrait d’ailleurs sans doute considérer une cataracte totale congénitale ou précoce comme une pathologie de l’ensemble du segment antérieur avec des conséquences qui dépassent le cristallin.
L’amélioration progressive de la technique opératoire et de la qualité des implants a permis de réduire progressivement l’âge d’implantation des enfants, mais l’indication dépend aussi de l’habitude et de l’expérience du chirurgien [1, 15]. Une implantation réalisée facilement et dans de bonnes conditions sera moins génératrice d’inflammation et de séquelles. Une courbe d’apprentissage est incontestable. Quel que soit l’âge de l’enfant, l’implantation ne sera réalisée que si les conditions locales sont bonnes, si possible en intercapsulaire et au cours d’une intervention non compliquée.
L’implantation n’est pas un complément simple de l’intervention mais elle est intégrée dans une stratégie thérapeutique de l’équipe ophtalmopédiatrique. On doit savoir dans chaque cas évaluer les bénéfices, notamment fonctionnels et les comparer aux risques liés au geste et à la présence de l’implant :
-
risques de l’implantation : iatrogénicité directe du geste (il y a davantage de manipulations intra-oculaires d’où plus d’inflammation, courbe d’apprentissage); biocompatibilité à long terme; risque d’évolution réfractive à intégrer dans la stratégie, différemment dans les cas bilatéraux et unilatéraux; opacifications secondaires plus fréquentes, accessibles à une réintervention;
-
les bénéfices sont avant tout réfractifs, en facilitant l’équipement optique, mais aussi la rééducation de l’amblyopie. Il est possible que la présence d’un implant, par sa rigidité, limite les risques de glaucome secondaire. Enfin, le moindre coÛt et la simplicité de la prise en charge ultérieure sont importants à prendre en compte, surtout dans les pays où l’accès aux soins est compliqué (et où les cataractes sont plus nombreuses).
Les implants en polymétachrylate de méthyle (PMMA) ont été utilisés en premier et leur biocompatibilité a été améliorée par l’adjonction d’un traitement de surface, hépariné ou fluoré, avec des résultats excellents. Des implants de taille adaptée aux sacs cristalliniens de l’enfant ont été commercialisés, simples à poser et avec une tolérance excellente à long terme. La pose d’implant PMMA a quasiment été abandonnée au profit des implants souples permettant de travailler sur une petite incision, plus sécurisée et moins astigmatogène. Les implants PMMA de chambre antérieure à appui angulaire sont déconseillés chez l’enfant en raison des risques pour l’angle et de leur proximité de la cornée. L’indication des implants PMMA à fixation irienne antérieure ou postérieure ou à fixation sclérale est discutée au cas par cas dans des situations spécifiques.
Les implants acryliques hydrophobes avec anses ouvertes sont donc recommandés actuellement. Ils semblent être les plus biocompatibles avec une rigidité suffisante pour résister à la rétraction du sac. Toutefois, jusqu’à ce jour, ils ne sont pas réalisés dans des tailles spécifiques à l’enfant. Il faut se méfier des filtres bleus qui diminueraient la biocompatibilité chez le jeune enfant. Les implants acryliques hydrophobes 3 pièces peuvent être utilisés en cas d’implantation secondaire dans le sulcus ciliaire.
Les implants multifocaux sont théoriquement séduisants. Ils sont franchement déconseillés pendant toute la période de croissance de l’œil en raison de l’évolution réfractive et de leur sensibilité au décentrement ou tant qu’une myopisation est en cours. En pratique, on peut les proposer au cas par cas après l’âge de 8 ans si la réfraction est stable et en l’absence de myopie (qui peut encore évoluer).
De nombreuses évolutions sont en discussion notamment pour tenter de limiter la complication la plus fréquente des cataractes infantiles qui est l’obstruction de l’axe visuel par la repousse de matériel cristallinien. On notera, par exemple, les discussions autour :
-
du rôle de la compression de la zone germinative équatoriale par les anses de l’implant ou un anneau;
-
de la meilleure fermeture du reliquat de sac par l’implant soit par luxation postérieure de l’optique, soit par implantation bag-in-the-lens dont les résultats sont excellents mais au prix d’une technique de pose complexe chez le nourrisson.
-
Ouverture cornéenne adaptée à la taille de l’implant ou de la cartouche.
-
Ouvertures capsulaires antérieures et postérieures calibrées (antérieure 6 mm, postérieure 4 mm), vérification de l’absence de vitré.
-
Injection de viscoélastique entre les feuillets capsulaires pour les ouvrir comme un pneu.
-
Glisser l’anse inférieure en intercapsulaire lors de l’injection, laisser déplier l’implant puis le faire tourner grâce à un crochet jusqu’à pouvoir luxer la deuxième anse entre les deux capsules. Ne pas hésiter à faire tourner l’implant afin de bien vérifier sa position.
-
Certains préfèrent implanter dans le sac, avant de réaliser la capsulotomie postérieure. Cette dernière est ensuite réalisée soit par voie antérieure en luxant l’optique de l’implant, soit par voie postérieure de vitrectomie
En présence d’un support capsulaire annulaire périphérique, cas le plus fréquent, on implante dans le sulcus. Les implants acryliques hydrophobes 3 pièces s’y glissent facilement. On prendra garde à réduire la puissance de l’implant de 1,5 D en raison de sa position plus antérieure. L’intervention s’accompagne en général de l’aspiration de masses résiduelles et d’un complément de vitrectomie antérieure. Elle se termine par une mise en myosis et éventuellement une iridectomie périphérique avant l’âge de 3 ans. En l’absence de support capsulaire, il n’existe pas de consensus. Certains préfèrent s’abstenir d’implanter. Il faut impérativement éviter les implants de chambre antérieure à fixation angulaire. Les implants à fixation irienne antérieure représentent sans doute le meilleur compromis actuel. Les implants à fixation irienne postérieure ou à fixation sclérale sont possibles, avec un risque de luxation postérieure ultérieure non négligeable
La puissance choisie dépend de plusieurs facteurs et, contrairement à l’adulte, on ne vise que rarement l’emmétropie immédiate chez l’enfant [16-20]. La puissance théorique permettant l’emmétropisation immédiate est calculée par les formules les plus adaptées : SRK T (Sanders, Retzlaff et Kraft, Théorique) et Holladay. Puisqu’on implante sur un œil en croissance, des abaques ont été mis au point afin de calculer statistiquement la puissance de l’implant permettant d’espérer une emmétropie à l’âge adulte. La puissance de l’implant posé est finalement adaptée en fonction de l’âge d’implantation et du type de cataracte.
Selon l’âge : de 0 à 3 mois = 60 % de SRK T; de 3 à 6 mois = 65 % ; de 6 à 12 mois = 70 % ; de 12 à 18 mois = 75 % ; de 18 à 24 mois = 80 % ; de 24 à 36 mois= 85 % ; de 36 à 60 mois = 90 % ; au-delà, 100 % .
Il faut aussi tenir compte d’une potentielle croissance oculaire « pathologique » , dont on peut se douter, en cas de longueur axiale insuffisante ou excessive pour l’âge, et qui entraînera une majoration ou minoration de la puissance.
Pour les cataractes bilatérales, on vise une emmétropie finale ou une légère myopie. En revanche, dans les cataractes unilatérales, une trop grande sous-correction initiale risque de mettre en péril la récupération visuelle. Il faut donc trouver un compromis entre le risque de myopisation secondaire et une qualité optique initiale optimale. Une sous-correction dosée est choisie en fonction des équipes.
En cas de myopisation excessive, un changement d’implant est envisageable une fois la réfraction stabilisée. Il s’agit, en général, de cas de myopisations axiales imprévisibles initialement; elles représenteraient 20 % des cas opérés avant l’âge de 1 an, surtout en cas de persistance du vitré primitif associée [21].
La chirurgie et les indications d’implantation dans les cataractes de l’enfant ont considérablement évolué ces dernières années, à la lumière des progrès de la chirurgie de l’adulte et de la réhabilitation visuelle de l’enfant. Le pronostic visuel de ces cataractes en est largement amélioré. Il s’agit d’une chirurgie d’autant plus spécialisée que l’enfant est jeune, s’intégrant dans une stratégie globale de traitement, décidée et organisée avant l’intervention par une équipe entraînée à ce type de prise en charge.
[1] Pediatric cataract surgery Baltimore: Lippincort Williams & Williams (2004).
[2] Lambert SR Treatment of congenital cataract Br J Ophthalmol 2004 ; 88 : 854-855
[3] Taylor D The Doyne Lecture. Congenital cataract : the history, the nature and the practice Eye 1998 ; 12 : 9-36
[4] Thouvenin D Management of infantile cataracts : surgical technics and choices in lens implantation J Fr Ophtalmol 2011 ; 34 : 198-202
[5] Wilson ME, Bartholomew LR, Trivedi RH Pediatric cataract surgery and intraocular lens implantation : practice styles and preferences of the 2001 ASCRS and AAPOS memberships J Cataract Refract Surg 2003 ; 29 : 1811-1820
[6] Trivedi RH, Peterseim MM, Wilson ME New techniques and technologies for pediatric cataract surgery Curr Opin Ophthalmol 2005 ; 16 : 289-293
[7] Roche O, Beby F, Orssaud C, et al. Cataracte congénitale J Fr Ophtalmol 2006 ; 29 : 443-455
[8] Parks MM Posterior lens capsulectomy during primary cataract surgery in children Ophthalmology 1983 ; 90 : 344-345
[9] Tassignon MJ, Gobin L, De Veuster I, Godts D Advantages of the bag-in-the-lens intraocular lens in pediatric cataract surgery J Fr Ophtalmol 2009 ; 32 : 481-487
[10] Dureau P Pathophysiology of zonular diseases Current Opinion in Ophthalmology 2008 ; 19 : 27-30
[11] Konradsen T, Kugelberg M, Zetterström C Visual outcomes and complications in surgery for ectopia lentis in children J Cataract Refract Surg 2007 ; 33 : 819-824
[12] Dureau P, de Laage de Meux P, Edelson C, Caputo G Iris fixation of foldable intraocular lenses for ectopia lentis in children J Cataract Refract Surg 2006 ; 32 : 1109-1114
[13] Infant Aphakia Treatment Study Group, Lambert SR, Lynn MJ, Hartmann EE, et al. Comparison of contact lens and intraocular lens correction of monocular aphakia during infancy: a randomized clinical trial of HOTV optotype acuity at age 4.5 years and clinical findings at age 5 years JAMA Ophthalmol 2014 ; 132 : 676-678
[14] Vasavada AR, Trivedi RH, Nath VC Visual axis opacification after AcrySof intraocular lens implantation in children J Cataract Refract Surg 2004 ; 30 : 1073-1081
[15] Thouvenin D, Lesueur L, Arne JL Implantation intercapsulaire dans les cataractes de l’enfant. Étude de 87 cas et comparaison à 88 cas sans implantation J Fr Ophtalmol 1995 ; 18 : 678-687
[16] Dahan E, Drusedeau M Choice of lens and dioptric power in pediatric pseudophakia J Cataract Refract Surg 1997 ; 23 : 618-623
[17] Thouvenin D, Nogue S, Fontes L, Arne JL Résultats fonctionnels à long terme du traitement des cataractes congénitales unilatérales opérées précocement J Fr Ophtalmol 2003 ; 26 : 562-569
[18] Wilson ME, Trivedi RH Eye growth after pediatric cataract surgery Am J Ophthalmol 2004 ; 138 : 1039-1040
[19] Depeyre C, Chapottot E, Arné JL, Thouvenin D Cataractes congénitales unilatérales opérées précocement : devenir réfractif à long terme J Fr Ophtalmol 2007 ; 30 : 457-462
[20] Birch EE, Stager DR Prevalence of good visual acuity following surgery for congenital unilateral cataract Arch Ophthalmol 1988 ; 106 : 40-43
[21] Thouvenin D, Lequeux L, Norbert O. IOL exchange for excessive myopic shift in 14 children operated for unilateral infantile cataract. 40th Annual Meeting of EPOS, Barcelona nov 6–8, 2014
A. Péchereau