Pathologie rétinienne dégénérative et/ou héréditaire
Coordonné par C. Hamel , G. Le Meur, I . Meunier
G. Le Meur
L'albinisme est une maladie génétique rare qui est caractérisée globalement par des déficits spécifiques du système visuel associés à un phénotype d'hypopigmentation, variable selon la perturbation de la production de mélanine. Le mot albinisme vient du latin albus qui signifie blanc. Historiquement, l'albinisme est avant tout un manque partiel ou total de pigmentation qui peut affecter la pigmentation des yeux uniquement ou des yeux et de la peau ou des cheveux, en entraînant un albinisme soit oculaire (ocular albinism [OA]) soit oculocutané (oculocutaneous albinism [OCA]) si une atteinte cutanée ou des phanères est adjointe. Des formes rares d'albinisme syndromiques existent : syndrome de Hermansky-Pudlak et syndrome de Chediak-Higashi (gène CHS1). C'est une pathologie importante pour l'ophtalmologiste, car c'est l'atteinte ophtalmologique qui fait souvent poser le diagnostic d'albinisme. Nous verrons successivement les diverses formes d'albinisme, la génétique en essayant de faire le lien avec la physiologie, la clinique des diverses formes, puis le traitement.
La prévalence de toute forme d'albinisme en Europe est de 1 pour 17 000 naissances [1]. Cette fréquence varie dans le monde avec une prévalence connue plus forte en Afrique en rapport avec des effets fondateurs et une consanguinité augmentée. L'albinisme est la cause d'environ 20 à 30 % des nystagmus sensoriels [2, 3] et serait à l'origine de 5 % des malvoyances dans le monde.
Il existe plusieurs formes d'albinismes : quatre formes principales d'OCA, trois formes mineures d'OCA nouvellement décrites et une forme d'OA.
L'albinisme oculocutané de type 1 ou OCA1 (Mendelian inheritance in man ou MIM : 203100) est le plus fréquent en Europe, puis par ordre de fréquence OCA2, OCA4, le type 3 étant rare [1]. Le type 1 est retrouvé dans 46 % des cas en Europe [4].
L'albinisme oculocutané de type 2 ou OCA2 (MIM : 203200) est le plus fréquent en Afrique avec des mutations spécifiquement retrouvées dans ce continent. Mais du fait de la migration historique entre nos deux continents, il n'est pas rare que ce type d'albinisme soit mis en évidence en Europe [1].
L'albinisme oculocutané de type 3 ou OCA3 (MIM : 203290) est rare en Europe [1].
L'albinisme oculocutané de type 4 ou OCA4 (MIM : 606574) est la forme la plus fréquente d'OCA au Japon où il représente 24 % des OCA. Dans le monde, sa prévalence est estimée à 1/100 000. La clinique est très hétérogène allant d'une hypopigmentation marquée à une coloration presque normale de la peau avec l'apparition, pour certains patients, d'une augmentation de la pigmentation au cours du temps et, pour d'autres, d'une stabilité de l'hypopigmentation au cours de leur vie [5].
L'albinisme oculocutané de type 5 ou OCA5 (MIM : 606574) n'a pas été décrit en Europe, il a juste été décrit dans une famille consanguine d'origine pakistanaise [6].
L'albinisme oculocutané de type 6 ou OCA6 (MIM : 609802) présente des signes cliniques ophtalmologiques classiques mais une atteinte très hétérogène des cheveux allant de l'hypopigmentation à une couleur normale. Sa prévalence dans le monde est actuellement inconnue mais en Europe elle est estimée à 1,25 % des cas d'OCA [7].
L'albinisme oculocutané de type 7 ou OCA7 est extrêmement rare et a été décrit dans deux publications : une famille de l'île de Féroé au Danemark et un patient lithuanien [8].
L'albinisme oculaire lié à l'X est un albinisme où seuls les signes oculaires sont présents. Il y a deux types d'albinisme oculaire décrits : le type 1, le plus fréquent, estimé à 1/50 000 (MIM : 300500) et le type 2 (MIM : 300600) où une forte myopie, une protanopie et une hespéranopie sont associées. Une troisième forme d'albinisme oculaire a été décrite; elle est associée à une surdité neurosensorielle (MIM : 300650).
Les gènes intervenant dans la survenue des albinismes codent pour des protéines qui jouent un rôle clé dans la biologie et la physiologie des cellules pigmentées où se produit la mélanine. Parmi ces gènes, certains codent pour : des enzymes clés de la mélanogenèse; des protéines « récepteurs-transporteurs-solutés » situées dans les mélanosomes, qui sont le compartiment subcellulaire lieu de production de la mélanine; des protéines impliquées dans la biogenèse des organelles associées aux lysosomes ou dans le trafic lysosomial (fig. 17-1) [8]. Actuellement, 19 gènes sont responsables des albinismes et des albinismes syndromiques (tableau 17-1). Malgré de nombreuses avancées dans la connaissance de la génétique des albinismes, environ 20 % des patients atteints restent actuellement sans diagnostic génétique [8].
La synthèse de la mélanine est réalisée dans les mélanosomes au sein des mélanocytes, suivie par le transfert des mélanosomes aux kératinocytes environnants qui vont ultérieurement transporter le pigment et éventuellement le dégrader. Au cours de leur maturation, les mélanosomes migrent de la région périnucléaire où ils sont produits, vers l'extrémité des dendrites. Les mélanocytes humains produisent deux types, chimiquement distincts, de mélanines : l'eumélanine, un pigment de couleur brun-noir et la phéomélanine, un pigment de couleur jaunerouge. Les principales étapes qui déterminent la pigmentation constitutive de la peau sont : la migration des mélanoblastes vers l'épiderme, leur survie et leur différenciation en mélanocytes, la densité des mélanocytes, l'expression et la fonction des constituants enzymatiques et structuraux des mélanosomes, la synthèse des différents types de mélanines, le transport des mélanosomes aux dendrites du mélanocyte, le transfert des mélanosomes aux kératinocytes et finalement la distribution des mélanines et leur dégradation au niveau de la peau. Au niveau des yeux, la mélanine est retrouvée au niveau du stroma irien, de l'épithélium irien, de l'épithélium pigmentaire de la rétine et de la choroïde. Au niveau de l'œil, elle a pour rôle d'empêcher la dispersion de la lumière dans l'œil, de détoxifier les cellules oculaires des produits oxydants qui proviennent de la dégradation des segments externes des photorécepteurs ou de la synthèse de la mélanine.
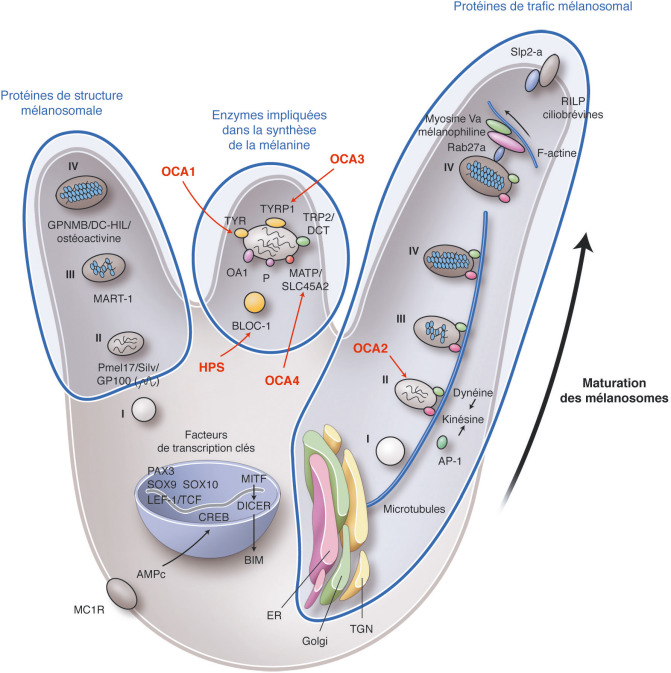
Fig. 17-1 Physiologie des mélanosomes et de la production de mélanine d'après Yamaguchi et Hearing.
Tableau 17-1 – Gènes et types d’albinismes.
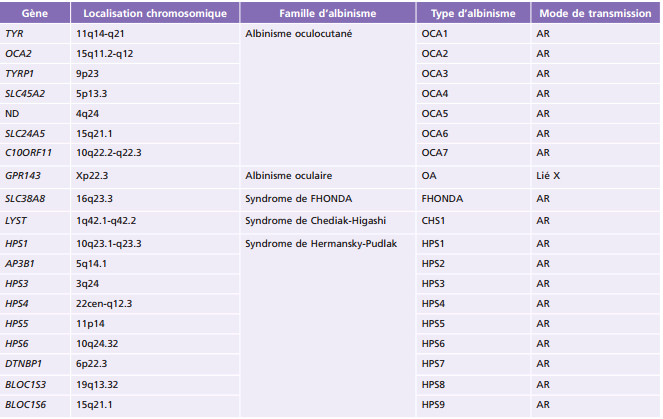
AR : autosomique récessive ; CHS : Chediak-Higashi syndrome ; FHONDA : foveal hypoplasia, optic nerve decussation defects and anterior segment dysgenesis ; HPS : Hermansky-Pudlak syndrome ; ND : non déterminé ; OA : ocular albinism ; OCA : oculocutaneous albinism.
Le gène TYR responsable de la forme OCA1, est composé de 5 exons couvrant environ 65 kb d'ADN génomique qui se compose de 529 acides aminés et qui code pour une protéine appelée tyrosinase (TYR). La protéine TYR est une enzyme contenant du cuivre qui catalyse la première des deux étapes de la voie de biosynthèse de la mélanine en réalisant la conversion de la tyrosine en L-dihydroxyphénylalanine (L-DOPA) et par la suite l'oxydation pour créer la dopaquinone [9]. Les mutations qui abolissent complètement l'activité de la tyrosinase donnent le sous-type OCA1A, alors que les mutations où persiste un peu d'activité enzymatique donnent la forme OCA1B [9].
Le gène OCA2 responsable d'OCA2, est composé de 24 exons sur 345 kb d'ADN génomique dans la région 15q11.2-q12. Ce gène code pour la protéine P ou pink-eyed dilution protein, de 838 acides aminés, qui se compose de 12 domaines transmembranaires. Cette protéine agit comme un précurseur de la synthèse de la mélanine au sein des mélanocytes. De plus, elle stabilise les protéines mélanosomales et leur trafic au sein du mélanosome [10].
Le gène TYRP1, responsable de la forme OCA3, est constitué de 8 exons et 7 introns, situés dans la région 9p23. Il code pour une protéine apparentée à la tyrosinase qui s'appelle tyrosinase 1 (Tyrp1) et qui semble être la protéine mélanosomale la plus abondante du mélanocyte. Tyrp1 partage une homologie de 40 à 52 % des acides aminés avec la tyrosinase (TYR). Tyrp 1 est impliqué dans le maintien de la structure des mélanosomes et affecte la prolifération des mélanocytes et la mort cellulaire. Tyrp1 est également impliqué dans la conversion de la L-tyrosine en DOPA avec faible rentabilité. Il est un cofacteur essentiel pour l'activité de la tyrosinase [9].
Le gène SLC45A2, responsable de OCA4, code pour une protéine de 530 amino-acides qui comprend 12 domaines transmembranaires appelée MATP (membrane-associated transporter protein). Elle jouerait un rôle dans le transport transmembranaire des mélanosomes [11].
Le gène responsable de la forme OCA5 n'a pas été encore déterminé. Il a été localisé en 4q24. Les études génétiques de liaison ont identifié 14 gènes candidats dans une région de 3,84 Mb située entre les marqueurs D4S421 et D4S2913 [8].
Le gène SLC24A5 responsable de la forme OCA6 est situé sur le chromosome 15q21.1. Il code pour une protéine NCKX5 échangeur de cations qui permettrait d'abaisser les organites Na par les transports couplés avec Ca et K au niveau du cytoplasme par transport des ions. Cette protéine serait située dans le mélanosome à un stade avancé ou dans le réseau trans-Golgi mais son interaction précise avec le Ca dans le mélanosome doit être encore déterminée (fig. 17-2) [12].
Le gène C10ORF11 est responsable de la survenue de l'OCA7 qui semble être de transmission autosomique récessive. Il code pour 198 acides aminés et jouerait un rôle dans la différenciation des mélanocytes [8].
Le gène GPR143, situé sur le chromosome X, est responsable de l'albinisme oculaire de type OA1. Il code pour une protéine transmembranaire située dans la membrane des mélanosomes. Cette protéine, joue le rôle de récepteur de la tyrosine, du L-DOPA et de la dopamine. De ce fait, elle joue un rôle important dans la biogenèse des mélanosomes.
Une entité à part vient d'être décrite, le syndrome FHONDA (foveal hypoplasia, optic nerve decussation defects and anterior segment dysgenesis), où sont retrouvées une hypoplasie fovéolaire, des anomalies de décussation chiasmatique et une dysgénésie du segment antérieur. Il est lié à une mutation du gène SLC38A8, situé dans la région 16q23.3-24.1. Le gène SLC38A8 code pour une protéine membre d'une famille de protéines SLC38, transporteurs d'acide aminé neutre couplé sodium (sodium-coupled neutral amino acid transporter [SNAT]) qui ont comme substrat préféré la glutamine [13].

Le premier signe ophtalmologique est souvent l'apparition d'un nystagmus précoce associé à une forte photophobie si l'atteinte est marquée. Le nystagmus est généralement horizontal, conjugué, avec une augmentation de la vitesse de la phase lente, de l'intensité du nystagmus et une modification de la forme d'onde lors des changements de direction du regard [14]. Le nystagmus est détecté entre 25-30 % et 90,9 % des cas d'albinisme [15, 16]. Le nystagmus peut être au premier plan des signes cliniques où alors il apparaît lors du début de la maturation de la macula vers l'âge de 3 à 4 mois. Parfois, ce nystagmus peut être plus fin et uniquement visible lors d'un examen à la lampe à fente ou par tomographie par cohérence optique (optical coherence tomography [OCT]) chez des patients examinés à cause d'une anomalie oculomotrice ou d'une limitation de l'acuité visuelle. Le nystagmus peut varier au cours du temps avec une aggravation à la fatigue, lors d'émotions fortes, lors d'éblouissement. Une position de torticolis accompagnant le nystagmus est fréquemment associée voire même un dodelinement de la tête. Ce torticolis permet au système visuel de trouver une « zone de calme » où le battement du nystagmus est plus faible, ce qui permet d'accroître les performances visuelles. C'est pourquoi cette position de calme est à respecter. De plus, il est nécessaire de l'expliquer aux parents afin qu'ils laissent l'enfant se positionner de manière la plus agréable et surtout la moins fatigante pour son système visuel. En lien avec ce nystagmus, il est noté que le temps de reconnaissance d'un mot isolé est augmenté chez les patients ayant un albinisme (versus groupe contrôle), il est en rapport avec une augmentation du nombre de fixations dont les durées sont plus courtes [17]. De plus, pour certains auteurs, à l'inverse d'autres [17], la vitesse de lecture est réduite de 18,8 % chez les personnes qui ont un albinisme par rapport à un groupe témoin [18].
La photophobie est variable en intensité et dans le temps. Il n'est pas rare que des parents d'enfants ayant un albinisme disent qu'après les premières années où celle-ci était vraiment handicapante, elle devient moins marquée. Cette photophobie peut se manifester par une position tête penchée en avant pour se protéger de la lumière, par un comportement plus alerte de l'enfant dans la pénombre.
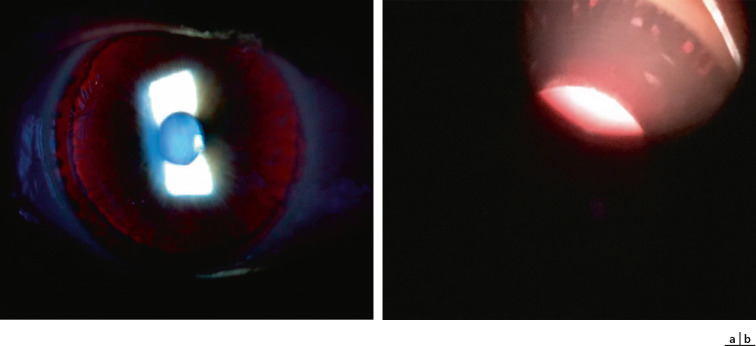
Fig. 17-3 Transillumination irienne : stade (a) et stade 1 (b).
La transillumination irienne, liée à l'hypopigmentation irienne, peut être variable et transitoire suivant les types d'albinismes. Il faut parfois faire un examen attentif à la lampe à fente afin de voir certaines transilluminations, par exemple dans certains types d'OCA2. Dans les cas typiques, un iris clair laisse passer la lumière avec une visibilité anormale du bord du cristallin et de la zonule (fig. 17-3a) ou, dans les formes mineures, des zones de transillumination de formes arrondies périphériques à la racine de l'iris sont visualisables (fig. 17-3b). Ces transilluminations périphériques peuvent disparaître au cours de la maturation de l'iris qui survient dans les premiers mois de vie. Une classification des transilluminations a été proposée par Summers :
- – stade 1 : transillumination périphérique ponctuée;
- – stade 2 : transillumination périphérique diffuse;
- – stade 3 : transillumination diffuse avec l'équateur du cristallin visible;
- – stade 4 : transillumination totale y compris le bord pupillaire.
L'épaisseur de l'iris en OCT est plus fine de 10,7 % chez les patients ayant un albinisme par rapport à un groupe témoin avec une corrélation avec le phénotype du patient, notamment le nystagmus, la coloration des cheveux et de la peau [19]. Ce défaut de pigmentation de l'iris mais aussi de pigmentation de la rétine est visible lors de l'examen du fond d'œil (fig. 17-4), et est responsable d'une photophobie importante chez les patients atteints d'albinisme. La pigmentation du fond d'œil est classée en trois stades selon la visibilité de la choroïde :
- – grade 1 : les vaisseaux choroïdiens sont facilement vus dans macula;
- – grade 2 : les vaisseaux choroïdiens sont moins distinctement vus à cause d'un épithélium pigmentaire rétinien qui reste toutefois translucide;
- – grade 3 : la macula est suffisamment opaque pour que les vaisseaux de la choroïde ne soient pas visibles [20].
Normalement, au cours du développement de la rétine, les cellules de la couche interne de la rétine migrent loin de la fovéa avec une migration des cônes dans la macula et un allongement des photorécepteurs. Ce processus est arrêté prématurément en cas d'albinisme, c'est pourquoi une hypoplasie fovéolaire est fréquemment retrouvée dans l'albinisme même si elle n'est pas pathognomonique. Cette hypoplasie peut être visible lors de l'examen du fond d'œil avec un reflet fovéolaire absent ou peu développé; en autofluorescence, elle apparaît sous la forme d'une absence de la tache noire fovéolaire mais elle est surtout facilement retrouvée lors de l'examen par OCT (fig. 17-4). C'est un des signes paracliniques essentiels et faciles à rechercher. Une analyse fine de la macula en mapping ne peut parfois pas être réalisée quand le nystagmus est trop important, toutefois une analyse en ligne permet le plus souvent en OCT de connaître le statut fovéolaire du patient qui a un albinisme. Une gradation de l'hypoplasie fovéolaire, quelle qu'en soit l'origine, par OCT a été proposée :
- – grade 1 : fosse fovéale peu profonde avec la présence d'un élargissement de la couche nucléaire externe (ou ONL pour outer nuclear layer) et d'un allongement du segment externe des photorécepteurs (ou OS pour outer segment);
- – grade 2 : classe 1, mais avec une absence de dépression fovéolaire;
- – grade 3 : grade 2 avec une absence d'allongement d'OS en plus;
- – grade 4 : grade 3 avec l'absence d'un élargissement d'ONL associée (fig. 17-5) [21].
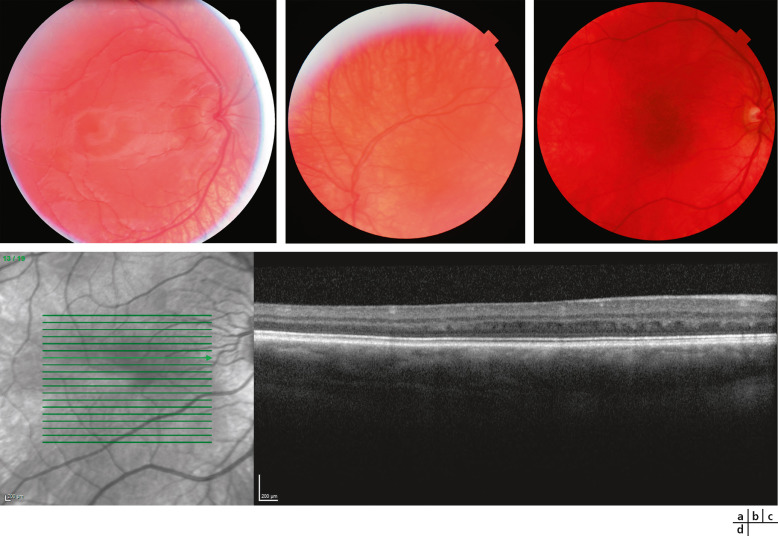
Fig. 17-4 a-d. Hypopigmentation rétinienne au fond d’oeil et hypoplasie fovéolaire à l’examen OCT.
Dans une étude menée en spectral-domain optical coherence tomography (SD-OCT) chez 44 patients albinos âgés entre 0 et 6 ans, l'épaisseur centrale maculaire de la rétine est sensiblement plus épaisse dans le groupe de l'albinisme que dans le groupe témoin (p < 0,0001) [22]. Cette majoration de l'épaisseur serait liée à l'étirement des photorécepteurs fovéolaires, notamment au niveau du segment interne de ces photorécepteurs fovéolaires [22]. En revanche, l'épaisseur de la rétine périfovéolaire est significativement diminuée dans l'albinisme aussi bien dans la couche interne que dans la couche externe de la rétine [22]. La présence de cercles concentriques maculaires lors de l'examen par infrared reflectance (IRR) en OCT est notée chez les patients ayant un albinisme. L'étiopathogénie de ce signe d'examen serait l'orientation concentrique des axones de la couche de Henlé autour de la fovéa en cas d'hypoplasie fovéale, alors que dans la fovéa normale, les axones des photorécepteurs seraient orientés radialement dans une configuration horizontalement symétrique [23]. Au niveau maculaire, Wolfson et al. ont mesuré une diminution du pigment maculaire lors de l'analyse par scanning laser ophthalmoscopy (SLO) chez 4 patients albinos qui avaient une hypoplasie fovéolaire [24], ce qui suggère que l'absence de dépression fovéolaire n'empêche pas la distribution du pigment maculaire. De plus, les études en optique adaptative ont permis de montrer qu'une orientation normale des cônes fovéolaires était possible malgré l'hypoplasie fovéolaire mais que la densité de ces cônes ainsi que leur allongement étaient aussi variables dans l'albinisme [25,26].
Les patients atteints d'albinisme présentent une anomalie de décussation des fibres temporales du nerf optique (fig. 17-6). Cette anomalie, connue depuis les années 1950, est à rechercher par des potentiels évoqués visuels (PEV) 5 voies, notamment lors de formes peu sévères de nystagmus. Cet examen permet de découvrir l'étiologie du nystagmus quand ce signe est présent, ce qui n'arrive qu'entre 66 % et 23 % des cas en fonction des formes d'albinisme [27]. En rapport avec cette particularité anatomique, une absence de vision stéréoscopique est fréquemment associée à l'albinisme, même en l'absence de strabisme [20, 27, 18].
Il est très fréquent dans l'albinisme comparé aux autres nystagmus congénitaux. Sa prévalence est décrite entre 53 à 90 % des patients atteints d'albinisme [29, 30]. Par ordre de fréquence, les strabismes lors de la fixation au loin sont en ésodéviation pour 55,5 % des cas, en exodéviation pour 23,8 % des cas associés ou non à un strabisme vertical pour 11,1 % avec sensiblement les mêmes résultats pour la fixation de près [30]. Une particularité anatomique oculomotrice a été décrite : les jonctions neuromusculaires semblent être moins nombreuses au niveau des muscles oculomoteurs chez les enfants ayant un albinisme comparés à un groupe témoin, comme si la présence d'un nystagmus induisait une adaptation de la voie finale commune pour se mettre dans une situation de moindre mouvement [31]. La présence d'un angle kappa positif est fréquemment retrouvée chez les patients ayant un albinisme dont l'origine serait liée, d'après McCafferty, à une fixation temporale maculaire notée lors de la visualisation de la rétine par imagerie [32]. La présence de cet angle kappa positif peut masquer une ésotropie ou alors majorer la sensation d'exotropie chez ces patients.
Toutes les amétropies peuvent être retrouvées dans les albinismes. L'équipe de Kurma retrouve, dans une étude faite chez des enfants vivants dans un milieu rural en Afrique, une sphère moyenne de – 3,26 D avec une médiane à – 1,5 D et des extrêmes allant de – 30,00 D à + 16 D [33]. Dans l'étude de Yaholam, l'amétropie la plus fréquemment retrouvée est l'astigmatisme avec un astigmatisme moyen de 2,1 D, présent dans 53 % des cas, quel que soit le type d'albinisme sauf en cas d'OCA1 où une forte hypermétropie (> 5 D) a été mise en évidence [16]. Mais ces règles pour les sphères ne semblent pas universelles et varient en fonction des études; par exemple, Kassmann décrit une association entre la myopie et OCA1 et entre l'hypermétropie et OCA2 [34]. En revanche, toutes les études s'entendent sur la présence d'un astigmatisme allant de 1,09 à 2,1 D [16, 33, 35]. L'acuité visuelle moyenne est comprise entre 20/320 et jusqu'à une acuité visuelle proche de la normale pour les formes mineures, selon les études, avec une acuité visuelle moyenne à 20/83 [20, 33, 35, 36]. Dans une étude de qualité de vie, menée chez 40 patients atteints d'albinisme, 50 % avaient une déficience visuelle moyenne et 50 % avaient une déficience visuelle de modérée à sévère [36]. La vision des couleurs est en général normale avec une sensibilité aux contrastes relativement bonne [33].
Fig. 17-5 Types d’hypoplasies fovéolaires d’après Thomas et al.
CCG : couche des cellules ganglionnaires ; CFO : couche des fibres optiques ; CNE : couche nucléaire externe ; CFNR : couche des fibres nerveuses rétiniennes ;
CNI : couche nucléaire interne (ou couche des noyaux des cellules bipolaires) ; CPE : couche plexiforme externe ; CPI : couche plexiforme interne ; EPR : épithélium pigmentaire rétinien ; MLE : membrane limitante interne ; SE : segment externe des photorécepteurs ; ZE : zone ellipsoïde.
Une hypopigmentation ou hypomélanose sera plus ou moins sévère. Elle est parfois difficile à discerner chez les Caucasiens au plus jeune âge ou dans les formes mimines et elle n'apparaît pas dans les formes oculaires. Dans les formes sévères, l'hypopigmentation est généralisée aux phanères et à la peau et est indépendante de la race : la peau est rose, les cheveux, les cils et les sourcils sont blanc neige (fig. 17-7). Dans les formes moins sévères, un certain degré de pigmentation peut apparaître avec la croissance comme dans les formes de type 2 d'albinisme oculocutané. Le manque de mélanine chez les personnes albinos les rend donc plus vulnérables aux rayons ultraviolets, ce qui fait qu'elles attrapent des coups de soleil plus rapidement que les personnes normalement pigmentées. Des expositions répétées sans protection peuvent malheureusement conduire à l'apparition de brÛlures de la peau, qui avec le temps peuvent provoquer un cancer de la peau. Le cancer de la peau est une cause majeure de morbidité et de mortalité chez les albinos qui développent les lésions précancéreuses et malignes à un plus jeune âge et souffrent de cancers avancés de la peau dans la troisième à la quatrième décennie de la vie notamment en Afrique [37]. La majorité des cancers cutanés dans les OCA sont des carcinomes épidermoïdes (75-88 % ), puis des carcinomes basocellulaires (9-23 % ), tandis que le mélanome malin est rare (1,3-3 % ) [37, 38]. Dans une étude récente, il ne semble pas y avoir de corrélation entre le nombre de lésions pigmentées (une centaine par patient) et l'âge des patients [39]. Le mélanome malin dans cette pathologie est rare : du fait de la grande sensibilité aux brÛlures de leur peau, ces patients sont peu exposés au soleil dans leur enfance.
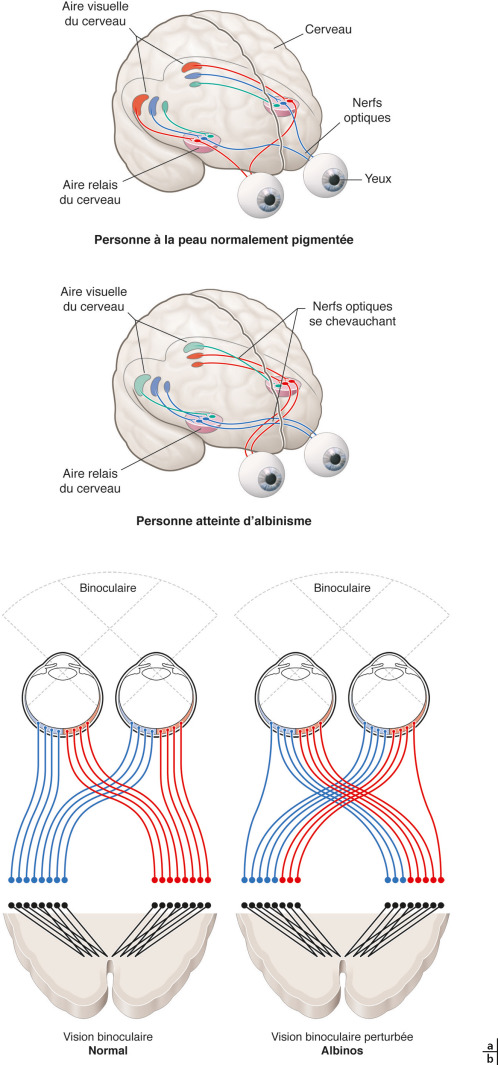
Fig. 17-6 a, b. Anomalie de décussation présente en cas d’albinisme.
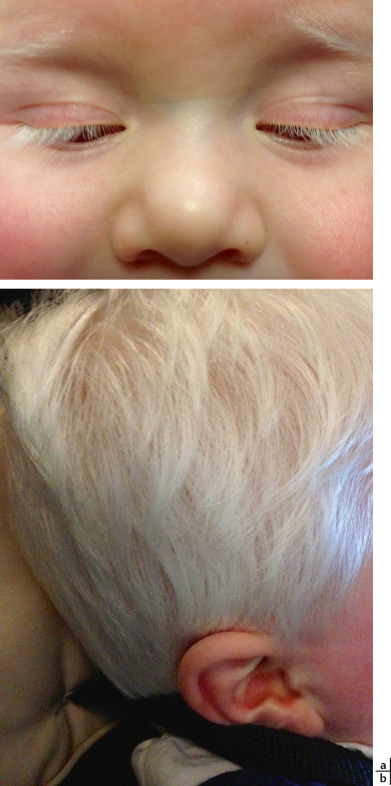
Fig. 17-7 a, b. Enfant avec un albinisme oculocutané.
Ce syndrome comprend un tableau clinique proche de l'albinisme oculocutané de type 2 auquel se rajoutent des anomalies du stockage des plaquettes avec l'apparition d'hémorragies lors de traumatismes mineurs (épistaxis, saignements après des interventions chirurgicales mineures). C'est pourquoi ce diagnostic différentiel de l'albinisme oculocutané est important à connaître. Il existe neuf types de syndrome d'Hermanski-Pudlak en fonction de l'atteinte génétique, qui ont le même phénotype clinique avec des degrés de sévérité variable, et de l'adjonction d'autres atteintes viscérales (tableau 17-1). Ces neuf gènes codent pour des protéines impliquées dans la biogenèse des organelles liées aux lysosomes (biogenesis of lysosome-related organelles complex [BLOC]) (fig. 17-1). Ces organelles blocs se retrouvent y compris dans les mélanosomes des ménalocytes et des granulés delta des plaquettes. La prévalence de ce syndrome est de 1/500 000 à 1/1 000 000 dans la population non portoricaine et de 1/1800 dans le nord de Porto Rico à cause d'un effet fondateur [40]. Chez 15 % des patients, une inflammation chronique de l'intestin s'associe au phénotype d'AOC [41]. Dans le type 2, une neutropénie est décrite [42]. Une fibrose pulmonaire peut être détectée chez les patients atteints de la forme de type 1, 2 et 4 et dont les premiers signes pulmonaires apparaissent vers 30 à 40 ans [43, 44].
Il associe des signes d'albinisme oculocutané, une immunodéficience et des problèmes neurologiques. Ce syndrome, de transmission autosomique récessive, est lié à une mutation du gène LYS dont la protéine intervient dans le trafic lysosomal [45]. La forme classique de SCH, qui représente plus de 80 % des cas, débute dans l'enfance et se caractérise par une immunodéficience et des infections récurrentes, une hémophagocytose et des changements pigmentaires affectant la peau et les cheveux et ressemblant à l'albinisme oculocutané. Une immunodéficience sévère et une lymphohistiocytose peuvent être retrouvées et sont souvent mortelles ou mettent la vie en jeu dans la première décennie [46]. L'atteinte neurologique peut se produire dans 10 % des cas infantiles. Cependant, environ 10 à 15 % des patients suivent une évolution clinique moins sévère : le SCH de l'adolescent ou le SCH de l'adulte. Dans ces formes atténuées ou après transplantation hématopoïétique de cellules souches dans les formes sévères, des signes neurologiques progressifs peuvent apparaître ou être présents lors du diagnostic : un déclin cognitif progressif, une démence, une neuropathie périphérique, des paralysies des nerfs crâniens, un syndrome parkinsonien, des anomalies de l'équilibre, une ataxie cérébelleuse ou des tremblements [46]. Les signes ophtalmologiques décrits dans ce syndrome sont, le plus souvent, un albinisme oculaire avec une hypoplasie fovéale et un nystagmus, mais une dégénérescence rétinienne pigmentaire a également été décrite [47]. Au niveau des atteintes neurologiques périphériques, une atteinte des nerfs optiques avec une atrophie optique secondaire, apparue vers l'âge de 35 ans, a été décrite dans une fratrie. Les signes cliniques ont été une perte de la vision colorée et des contrastes, l'apparition d'un scotome central, d'une pâleur temporale du nerf optique avec une atteinte à l'OCT retinal nerve fiber layer (RNFL) [48]. Ce syndrome doit être suspecté chez les enfants atteints d'albinisme qui présentent des infections à répétition afin de pouvoir mettre le plus tôt en place un traitement et une prise en charge adaptés [46].
Actuellement, aucun traitement n'a d'autorisation de marché dans cette indication, mais une prise en charge adaptée doit être proposée aux patients ayant un albinisme oculocutané et à leur famille.
La correction des amétropies doit être envisagée et être réalisée dès le plus jeune âge comme lors de toute prise en charge d'un nystagmus congénital. Le port de la correction optique a été montré comme améliorant l'acuité visuelle d'environ 2 lignes dans une étude réalisée chez 35 patients [35]. Schulze Schwering trouve aussi la même amélioration d'acuité visuelle quand la correction optique est portée pour la moitié des 120 patients atteints d'albinisme oculocutané de type 2 examinés (surtout les patients qui avaient une myopie de moyenne à modérée) [33]. La plus grande amélioration a été réalisée chez les patients myopes entre – 1 et – 7,5 D. Une amélioration significative de l'angle du strabisme lorsque les verres ont été portés a également été retrouvée dans l'étude d'Anderson [35]. Le port de lunettes teintées doit être proposé quand la photophobie est invalidante mais en faisant attention que la teinte ne soit pas trop foncée notamment à l'intérieur. Les chapeaux et casquettes sont aussi un bon moyen de protéger les yeux du soleil. En général, les postures anormales de la tête ne semblent pas être améliorées par des lunettes. Le port de lentilles de contact peut être proposé, dès que possible, afin d'améliorer la qualité de la correction optique. Si la position de torticolis est importante ou à l'origine de cervicalgies trop importantes, il faudra proposer une chirurgie oculomotrice mais en attendant que l'enfant mature son système visuel, car les positions de tête peuvent varier dans l'enfance. Il faudra donc attendre que l'enfant n'ait plus de système de surcorrection ou cache, qu'il ait choisi son œil directeur avant d'envisager une chirurgie oculomotrice qui vise à améliorer la position de torticolis. De même, la position tête penchée en avant à cause de la photophobie sera bien à différencier de la position de calme du nystagmus. L'hypopigmentation de la peau doit être protégée contre l'exposition au soleil et les coups de soleil, par l'utilisation adéquate des vêtements longs, de tissage suffisamment dense, au besoin de vêtements anti-ultraviolets, par des chapeaux ainsi que par des crèmes solaires efficaces (indice 50) avec un respect des applications fréquentes. Un dossier MDA (maison de l'autonomie) pour une prise en charge en S3AIS pour la scolarité peut être effectué si l'acuité visuelle est inférieure à 4/10. Enfin, l'association Genespoir est un recours pour les patients et leur famille.
Dans les syndromes de Hermanski-Pudlak, il faut éviter l'aspirine et les anti-inflammatoires non stéroïdiens à cause du risque hémorragique.
[1] Martinez-Garcia M, Montoliu L Albinism in Europe J Dermatol: ( 2013 ) :40: 319-324
[2] Sarvananthan N, Surendran M, Roberts EO The prevalence of nystagmus : the Leicestershire nystagmus survey Invest Ophthalmol Vis Sci: ( 2009 ) :50: 5201-5206
[3] Lorenz B, Gampe E Analysis of 180 patients with sensory defect nystagmus (SDN) and congenital idiopathic nystagmus (CIN) Klin Monatsblätter Für Augenheilkd: ( 2001 ) :218: 3-12
[4] Rooryck C, Morice-Picard F, Elçioglu NH Molecular diagnosis of oculocutaneous albinism : new mutations in the OCA1-4 genes and practical aspects Pigment Cell Melanoma Res: ( 2008 ) :21: 583-587
[5] Straniero L, Rimoldi V, Soldà G Two novel splicing mutations in the SLC45A2 gene cause oculocutaneous albinism type IV by unmasking cryptic splice sites J Hum Genet: ( 2015 ) :60: 467-471
[6] Kausar T, Bhatti MA, Ali M OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24 Clin Genet: ( 2013 ) :84: 91-93
[7] Veniani E, Mauri L, Manfredini E Detection of the first OCA6 Italian patient in a large cohort of albino subjects J Dermatol Sci: ( 2016 ) :81: 208-209
[8] Montoliu L, Gr0nskov K, Wei AH Increasing the complexity : new genes and new types of albinism Pigment Cell Melanoma Res: ( 2014 ) :27: 11-18
[9] Kamaraj B, Purohit R Mutational analysis of oculocutaneous albinism : a compact review BioMed Res Int: ( 2014 ) :2014: 905472-
[10] Park S, Morya VK, Nguyen DH Unrevealing the role of P-protein on melano-some biology and structure, using siRNA-mediated down regulation of OCA2 Mol Cell Biochem: ( 2015 ) :403: 61-71
[11] Kamaraj B, Purohit R Mutational analysis on membrane associated transporter protein (MATP) and their structural consequences in oculocutaeous albinism type 4 (OCA4)-a molecular dynamics approach J Cell Biochem: ( 2016 ) :117: 2608-2619
[12] Jalloul AH, Rogasevskaia TP, Szerencsei RT, Schnetkamp PPM Array J Biol Chem: ( 2016 ) :291: 13113-13123
[13] Poulter JA, Al-Araimi M, Conte I, van Genderen MM Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism Am J Hum Genet: ( 2013 ) :93: 1143-1150
[14] Papageorgiou E, McLean RJ, Gottlob I Nystagmus in childhood Pediatr Neonatol: ( 2014 ) :55: 341-351
[15] Kumar A, Gottlob I, McLean RJ Clinical and oculomotor characteristics of albinism compared to FRMD7 associated infantile nystagmus Invest Ophthalmol Vis Sci: ( 2011 ) :52: 2306-2313
[16] Yahalom C, Tzur V, Blumenfeld A Refractive profile in oculocutaneous albinism and its correlation with final visual outcome Br J Ophthalmol: ( 2012 ) :96: 537-539
[17] Dysli M, Abegg M Nystagmus does not limit reading ability in albinism PloS One: ( 2016 ) :11: e0158815
[18] Barot N, McLean RJ, Gottlob I, Proudlock FA Reading performance in infantile nystagmus Ophthalmology: ( 2013 ) :120: 1232-1238
[19] Sheth V, Gottlob I, Mohammad S Diagnostic potential of iris cross-sectional imaging in albinism using optical coherence tomography Ophthalmology: ( 2013 ) :120: 2082-2090
[20] Summers CG Albinism : classification, clinical characteristics, and recent findings Optom Vis Sci: ( 2009 ) :86: 659-662
[21] Thomas MG, Kumar A, Mohammad S Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology: ( 2011 ) :118: 1653-1660
[22] Lee H, Purohit R, Sheth V Retinal development in albinism : a prospective study using optical coherence tomography in infants and young children Lancet Lond Engl: ( 2015 ) :385: S14
[23] Cornish KS, Reddy AR, McBain VA Concentric macular rings sign in patients with foveal hypoplasia JAMA Ophthalmol: ( 2014 ) :132: 1084-1088
[24] Wolfson Y, Fletcher E, Strauss RW, Scholl HPN Evidence of macular pigment in the central macula in albinism Exp Eye Res: ( 2016 ) :145: 468-471
[25] Wilk MA, McAllister JT, Cooper RF Relationship between foveal cone specialization and pit morphology in albinism Invest Ophthalmol Vis Sci: ( 2014 ) :55: 4186-4198
[26] McAllister JT, Dubis AM, Tait DM Arrested development : high-resolution imaging of foveal morphology in albinism Vision Res: ( 2010 ) :50: 810-817
[27] Lee KA, King RA, Summers CG Stereopsis in patients with albinism : clinical correlates J AAPOS: ( 2001 ) :5: 98-104
[28] Huurneman B, Boonstra FN Monocular and binocular development in children with albinism, infantile nystagmus syndrome, and normal vision Strabismus: ( 2013 ) :21: 216-224
[29] Brodsky MC, Fray KJ The prevalence of strabismus in congenital nystagmus : the influence of anterior visual pathway disease J AAPOS: ( 1997 ) :1: 16-19
[30] Merrill K, Hogue K, Downes S Reading acuity in albinism : evaluation with MNREAD charts J AAPOS: ( 2011 ) :15: 29-32
[31] McLoon LK, Willoughby CL, Anderson JS Abnormally small neuromuscular junctions in the extraocular muscles from subjects with idiopathic nystagmus and nystagmus associated with albinism Invest Ophthalmol Vis Sci: ( 2016 ) :57: 1912-1920
[32] McCafferty BK, Wilk MA, McAllister JT Clinical insights into foveal morphology in albinism J Pediatr Ophthalmol Strabismus: ( 2015 ) :52: 167-172
[33] Schulze Schwering M, Kumar N, Bohrmann D Refractive errors, visual impairment, and the use of low-vision devices in albinism in Malawi Graefes Arch Clin Exp Ophthalmol: ( 2015 ) :253: 655-661
[34] Käsmann B, Ruprecht KW Might the refractive state in oculocutaneous albino patients be a clue for distinguishing between tyrosinase-positive and tyrosinase-negative forms of oculocutaneous albinism? Ger J Ophthalmol: ( 1996 ) :5: 422-427
[35] Anderson J, Lavoie J, Merrill K Efficacy of spectacles in persons with albinism J AAPOS: ( 2004 ) :8: 515-520
[36] Kutzbach BR, Merrill KS, Hogue KM Evaluation of vision-specific quality-of-life in albinism J AAPOS: ( 2009 ) :13: 191-195
[37] Kiprono SK, Chaula BM, Beltraminelli H Histological review of skin cancers in African Albinos : a 10-year retrospective review BMC Cancer: ( 2014 ) :14: 157
[38] Mabula JB, Chalya PL, Mchembe MD Skin cancers among Albinos at a University teaching hospital in Northwestern Tanzania : a retrospective review of 64 cases BMC Dermatol: ( 2012 ) :12: 5
[39] Van der Westhuizen G, Beukes CA, Green B A histopathological study of melanocytic and pigmented skin lesions in patients with albinism J Cutan Pathol: ( 2015 ) :42: 840-846
[40] Witkop CJ, Nuñez Babcock M, Rao GH Albinism and Hermansky-Pudlak syndrome in Puerto Rico Bol Asoc Médica P R: ( 1990 ) :82: 333-339
[41] Hussain N, Quezado M, Huizing M Intestinal disease in Hermansky-Pudlak syndrome : occurrence of colitis and relation to genotype Clin Gastroenterol Hepatol: ( 2006 ) :4: 73-80
[42] Wenham M, Grieve S, Cummins M Two patients with Hermansky Pudlak syndrome type 2 and novel mutations in AP3B1 Haematologica: ( 2010 ) :95: 333-337
[43] Bin Saeedan M, Faheem Mohammed S, Mohammed TL Hermansky-Pudlak syndrome : high-resolution computed tomography findings and literature review Curr Probl Diagn Radiol: ( 2015 ) :44: 383-385
[44] Vicary GW, Vergne Y, Santiago-Cornier A Pulmonary fibrosis in Hermansky-Pudlak syndrome Ann Am Thorac Soc: ( 2016 ) :13: 1839-1846
[45] Nagle DL, Karim MA, Woolf EA Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome Nat Genet: ( 1996 ) :14: 307-311
[46] Lozano ML, Rivera J, Sánchez-Guiu I, Vicente V Towards the targeted management of Chediak-Higashi syndrome Orphanet J Rare Dis: ( 2014 ) :9: 132
[47] Sayanagi K, Fujikado T, Onodera T, Tano Y Chediak-Higashi syndrome with progressive visual loss Jpn J Ophthalmol: ( 2003 ) :47: 304-306
[48] Desai N, Weisfeld-Adams JD, Brodie SE Optic neuropathy in late-onset neurodegenerative Chediak-Higashi syndrome Br J Ophthalmol: ( 2016 ) :100: 704-707
I . Meunier, B. Puech, X. Zanlonghi , S. Defoort-Dhel lemmes, C. Hamel
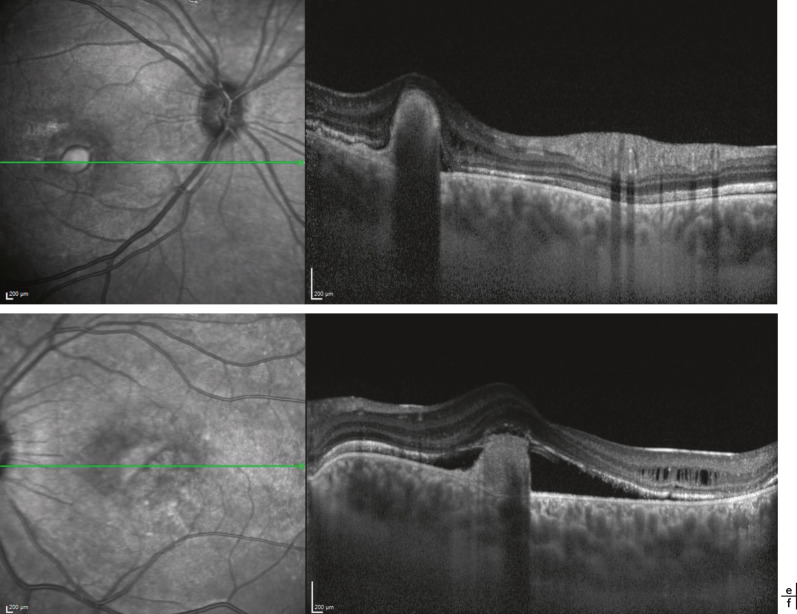
C'est la maculopathie héréditaire la plus fréquente; les taches flavimaculées, bien qu'inconstantes, en sont le signe majeur. Nous aborderons uniquement la forme juvénile et récessive où la macula peut paraître initialement peu remaniée malgré une acuité visuelle diminuée [1, 2]. Cette baisse d'acuité visuelle est précoce, le plus souvent avant l'âge de 10 ans, bilatérale et rapidement évolutive. Les anomalies de la vision des couleurs, rarement évoquées par l'enfant, sont en rapport avec la sévérité de la baisse d'acuité visuelle. La photophobie est plus tardive. Les difficultés d'adaptation à l'obscurité sont rarement rapportées et une héméralopie ira contre le diagnostic de maculopathie de Stargardt. Une héméralopie oriente alors vers une dystrophie mixte de type cônes-bâtonnets.
Les figures 17-8 et 17-9 récapitulent les différents aspects évolutifs du fond d'œil et de l'autofluorescence. Aux stades précoces, la macula peut paraître « normale » , ce qui peut conduire à un retard ou à une erreur de diagnostic. La perte de l'hypo-autofluorescence fovéolaire ou son étirement (oblongue et horizontale en « œil de bœuf » ) sont évocateurs à ces stades. Il en est de même d'une lésion maculaire très hétérogène composée de zones hypo- et hyper-autofluorescentes sans liseré périfovéolaire. Ce dernier aspect est plus en faveur d'une dystrophie des cônes ou d'une dystrophie mixte. Les taches flavimaculées, mieux visibles en autofluorescence, sont absentes, discrètes et limitées à l'aire maculaire ou diffuses et allant au-delà des vaisseaux temporaux avec un aspect très spécifique de fundus flavimaculatus. Les différents stades évolutifs en OCT spectral (B) sont présentés dans la figure 17-10..
Dans ces formes juvéniles, l'atrophie géographique est tardive, n'apparaissant que chez l'adulte [3]. La présence d'une telle atrophie chez l'enfant orientera vers d'autres pathologies telles un syndrome de Bardet-Biedl avec des réponses issues des cônes et des bâtonnets diminuées ou encore une céroïde lipofuscinose avec un aspect électro-négatif de l'électrorétinogramme (ERG).
L'ERG grand champ est obligatoire, y compris dans les formes typiques. Selon la classification de Lois, l'ERG est normal dans la majorité des cas, éliminant dès lors une dystrophie des cônes. L'ERG est plus rarement altéré avec soit une diminution des réponses issues des cônes, soit une diminution des réponses issues des cônes et des bâtonnets. Un risque de perte de la vision périphérique à moyen ou long terme est rapporté dans ces maculopathies de Stargardt avec un ERG altéré [4].
Depuis le développement du SD-OCT, l'ERG multifocal est rarement indiqué. Il peut être utile aux stades précoces où le fond d'œil paraît « normal » et où l'ERG grand champ est non altéré. On retrouve alors une baisse ou une absence de pic fovéolaire avec une diminution des réponses sur le premier anneau.
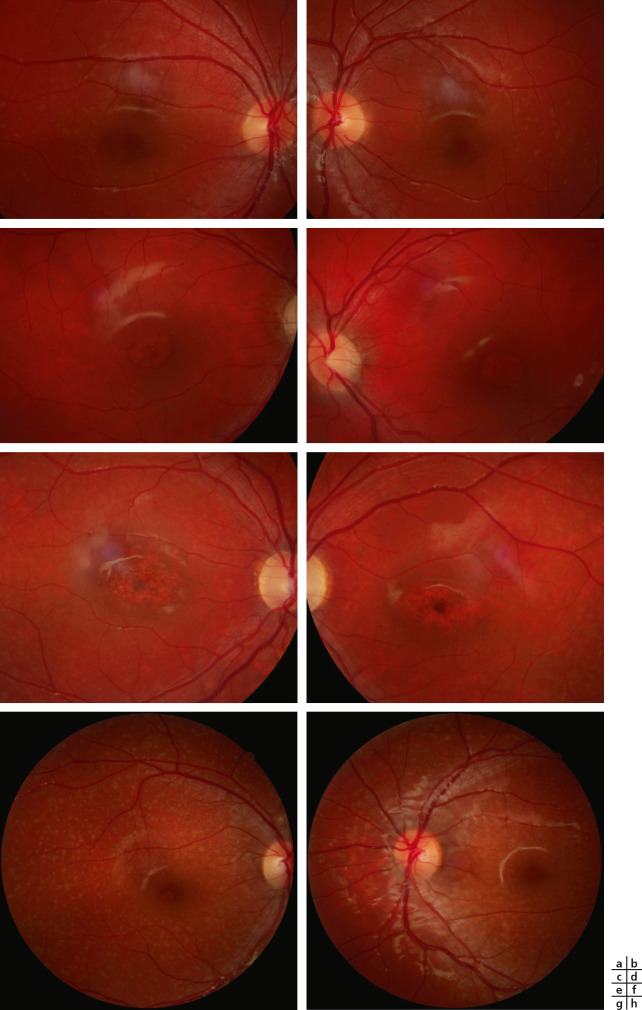
Fig. 17-8 Imagerie en couleurs dans la maculopathie juvénile de Stargardt.
a, b. La macula paraît normale malgré une acuité visuelle de 2/10 aux deux yeux chez un enfant de 12 ans. Noter l’absence de taches flavimaculées. c, d. Garçon âgé de 9 ans et demi avec une acuité visuelle de : 0,4 ; P5 oeil droit et 0,2 ; P6 oeil gauche. Les remaniements maculaires restent discrets malgré la baisse de l’acuité visuelle. e, f. Garçon de 9 ans avec une acuité visuelle de : 0,16 ; P6 à l’oeil droit et 2/10 ; P5 à l’oeil gauche. Le reflet fovéolaire est altéré avec des remaniements maculaires ovalaires d’axe horizontal. Pas de taches flavimaculées satellites visibles. Le calibre des vaisseaux rétiniens est conservé. g, h. Enfant âgé de 9 ans. Perte du reflet fovéolaire aux deux yeux avec de discrètes taches flavimaculées diffuses reproduisant un aspect de fundus flavimaculatus. L’acuité initiale est de 10/10, mais elle chutera à 0,16 aux deux yeux en 14 mois.
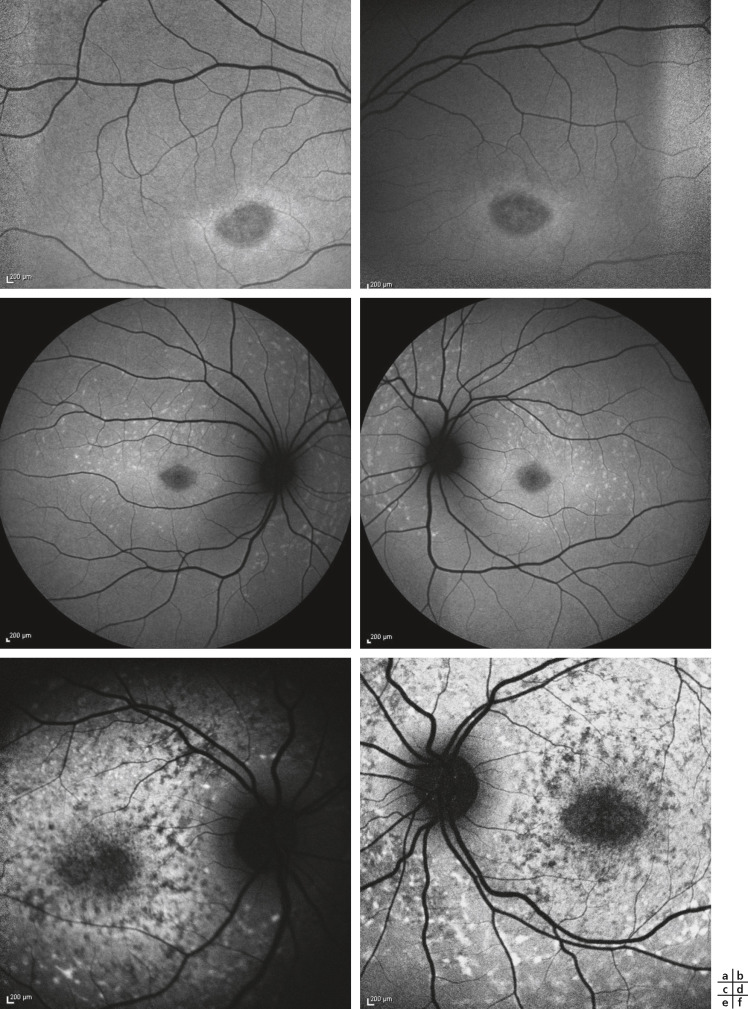
Fig. 17-9 Maculopathie juvénile de Stargardt, différents aspects en autofluorescence.
Trois éléments sont à analyser : la macula, les taches et la zone péripapillaire. Cette imagerie permet de détecter l’atteinte centrale au stade « infraclinique » : l’autofluorescence de la macula est inhomogène avec de multiples petites plages hypo-autofluorescentes, alors que le fond d’oeil semble normal ou peu remanié en biomicroscopie (a, b). Les taches flavimaculées sont hyper-autofluorescentes (lipofuscine) et plus nombreuses que sur les clichés en couleurs notamment pour les clichés c, d à comparer aux clichés en couleurs du même patient (clichés a et b de la fig. 17-8). La zone péripapillaire (e, f) est indemne, quel que soit le stade évolutif, sans lésions hypo-autofluorescentes (atrophie) ou hyper-autofluorescentes (taches flavimaculées).
Ces difficultés sont notées aux stades précoces ou en l'absence de taches flavimaculées. Il est alors important d'examiner les parents et les frères et sœurs. Classiquement, la pathologie étant récessive, seuls les frères ou sœurs peuvent présenter une maculopathie, avec parfois un aspect plus typique chez l'un d'entre eux. Les principaux diagnostics différentiels sont, chez l'enfant, un syndrome de Bardet-Biedl ou une céroïde lipofuscinose confirmant le rôle essentiel de l'ERG grand champ.
Le pronostic visuel est réservé avec une acuité visuelle finale variant de 1/20 à 2/10 avec présence d'un scotome central. La plupart des enfants ayant une forme purement centrale parviennent à suivre une scolarité classique. Les complications néovasculaires sont ici exceptionnelles. Il est essentiel que l'enfant se protège de la lumière avec des verres filtrants sur des montures couvrantes et le port de chapeau ou casquette. Il ne faut pas supplémenter l'enfant en vitamine A qui a un effet toxique. Chez l'adolescent, le tabac est à éviter (privilégier un environnement non fumeur).
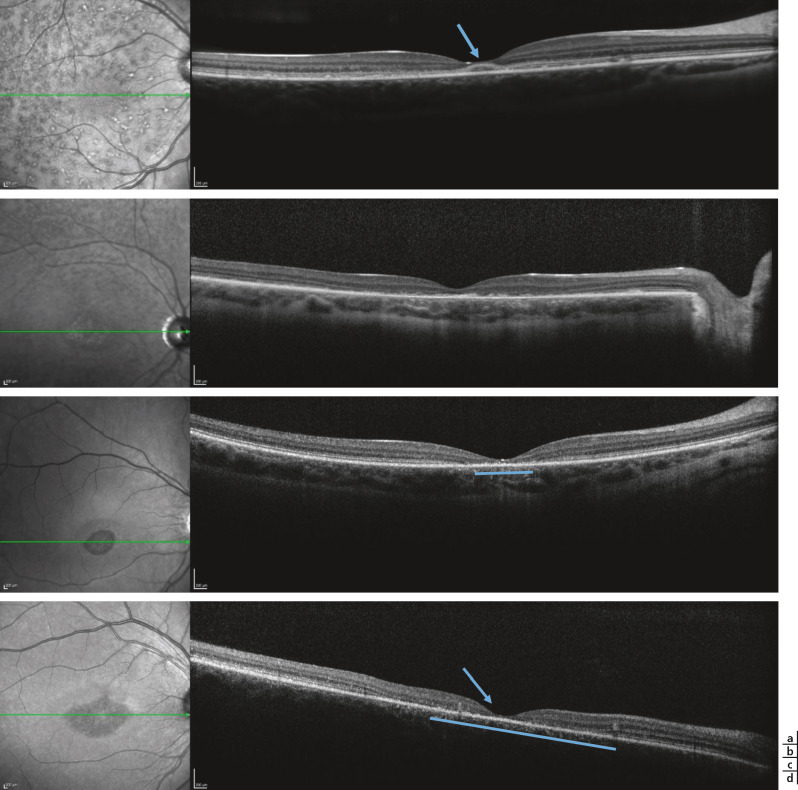
Fig. 17-10 Maculopathie juvénile de Stargardt, imagerie en SD-OCT mode B.
Les aspects sont variables mais ont une topographie caractéristique fovéomaculaire. On peut observer : un aspect épaissi granité (a, flèche et b) ou à l’inverse des discontinuités de la ligne ellipsoïde (c, d : dans la zone soulignée par la barre bleue), des anomalies de réflectivité ou à l’inverse un amincissement de la couche nucléaire externe en rapport avec la perte des photorécepteurs (c, d), puis ultérieurement, l’amincissement de la rétine. Les taches flavimaculées apparaissent comme des éléments hyperréflectifs en regard de l’épithélium pigmentaire ou sus-jacentes, détachées de l’épithélium pigmentaire (d, flèche).
Il est indispensable de confirmer l'implication du gène ABCA4 (ATP Binding Cassette Subfamily A Member 4) en vue d'une thérapie à venir. Certaines mutations sont introniques, trois d'entre elles sont actuellement testées. Les grands réarrangements (délétions, duplications) seront recherchés dans un deuxième temps. Si au terme d'une analyse complète (exons, mutations introniques, réarrangements), deux mutations bialléliques (une mutation sur chaque copie, l'une portée par le père, l'autre par la mère) sont identifiées, le diagnostic est alors génétiquement prouvé. Si une seule mutation est identifiée, le diagnostic reste probable à condition que le tableau clinique et l'évolution soient compatibles. Si aucune mutation n'est identifiée, alors le diagnostic de Stargardt est à reconsidérer. Des essais de thérapie génique et pharmacologique sont en cours. Par ailleurs, la forme dominante, similaire sur le plan clinique, est exceptionnelle et liée à des mutations hétérozygotes (mutation sur un seul allèle) dans le gène PRPH2 ou, plus rarement, dans le gène ELOVL4.
Initialement décrite par Adams en 1883, revisitée d'un point de vue génétique par Best en 1905 (transmission autosomique dominante), la maladie de Best (juvenile vitelliform macular disease) est caractérisée par la présence de dépôt(s) vitellin(s) autofluorescent(s), dont la séquence évolutive est stéréotypée, de l'apparition à la fragmentation du matériel jusqu'à sa résorption [5–7]. Les dépôts, uniques ou multiples, maculaires ou extramaculaires, peuvent être à des stades évolutifs différents, d'un œil à l'autre ou au sein du même œil. Cette asymétrie, tant de la taille des lésions que de leur stade, est particulièrement évocatrice des dystrophies vitelliformes qui sont les seules maculopathies héréditaires unilatérales ou bilatérales asymétriques.
La maladie de Best est découverte dans deux cas sur trois avant l'âge de 20 ans, le plus souvent avant l'âge de 10 ans. Contrairement à la maladie de Stargardt et aux dystrophies des cônes, la photophobie et les anomalies de vision des couleurs sont discrètes. De plus, les patients ne sont symptomatiques qu'aux stades de fragmentation et de résorption du matériel. Le matériel peut apparaître au deuxième œil avec un retard de plus de 10 ans.
Les figures 17-11 et 17-12 résument les différents aspects du fond d'œil et de l'autofluorescence. Aux stades prévitelliformes et vitelliformes, l'acuité visuelle est conservée. Dans le cas contraire, on recherchera une autre cause. Le matériel est autofluorescent (lipofuscine). L'autofluorescence est homogène ou inhomogène en fonction du stade d'évolution. Les petites zones hypo-autofluorescentes correspondent aux altérations de l'épithélium pigmentaire ou à l'atrophie choriorétinienne. Le matériel vitellin reproduit une lésion bombée hyperréflective en dessous de la ligne hyperréflective des photorécepteurs (fig. 17-13). Cependant, il est fréquent d'observer au stade de fragmentation des différences de réflectivité du matériel avec apparition de zones sombres optiquement vides reproduisant un aspect de faux décollement séreux rétinien. Ce dernier aspect de pseudo-décollement associé à du matériel ne doit pas faire évoquer une complication néovasculaire. Seules les modifications exsudatives intrarétiniennes sont un signe de néovascularisation.
Un rapport d'Arden anormal apporte un argument décisif au diagnostic de maladie de Best de l'enfant. Cette baisse du rapport d'Arden est liée à la diminution du passage transépithélial du chlore secondaire à l'altération fonctionnelle de la bestrophine. L'électro-oculogramme (EOG) nécessite la coopération de l'enfant qui doit accepter les contraintes de l'examen et pouvoir regarder à droite puis à gauche avec régularité. L'ERG n'est indiqué que dans les formes avec dépôts vitellins multiples ou dans les formes sévères. L'altération de l'ERG oriente dès lors vers une bestrophinopathie, forme rare récessive (dépôts multiples maculaires, rapport d'Arden diminué, parents non atteints et présence d'une mutation sur l'allèle paternel et sur l'allèle maternel).
Fig. 17-11 Imagerie en couleurs dans la maladie de Best juvénile.
Stades prévitellin (a, b) et vitellin (c, d) : le matériel est jaunâtre et homogène avec un aspect caractéristique d’oeuf sur le plat. L’acuité est ici conservée. Stade de fragmentation avec (e, f) ou sans hypopion (g, h). Aux stades suivants, le matériel se fragmente et l’acuité visuelle peut dès lors baisser. Stade de résorption du matériel avec des lésions de fibrose (i, j). La fibrose est un mode évolutif classique des néovaisseaux sur matériel.
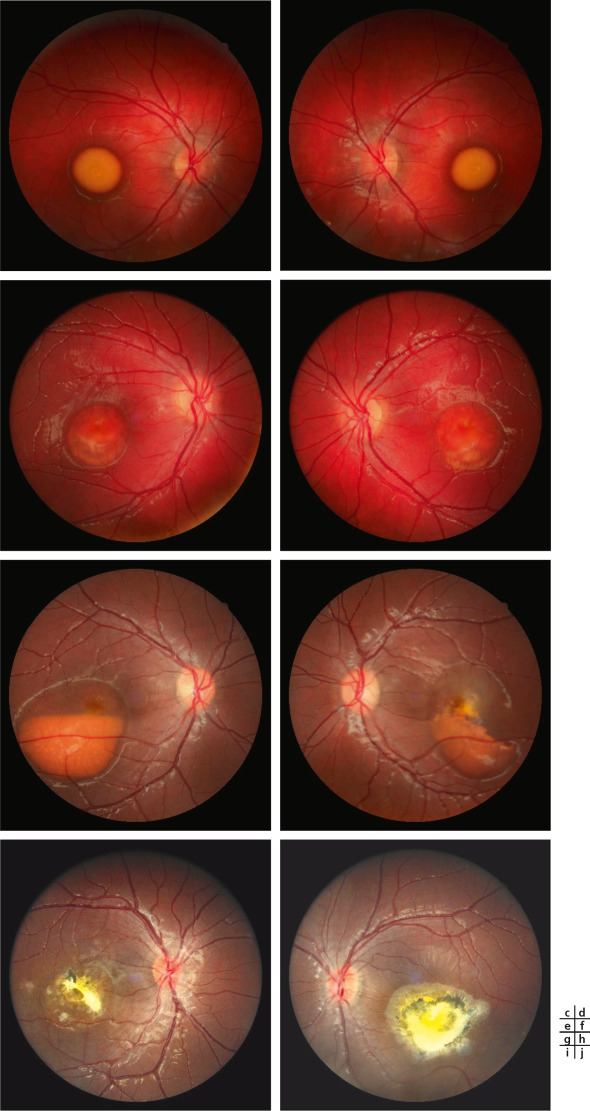
Fig. 17-11 Imagerie en couleurs dans la maladie de Best juvénile. (Suite)
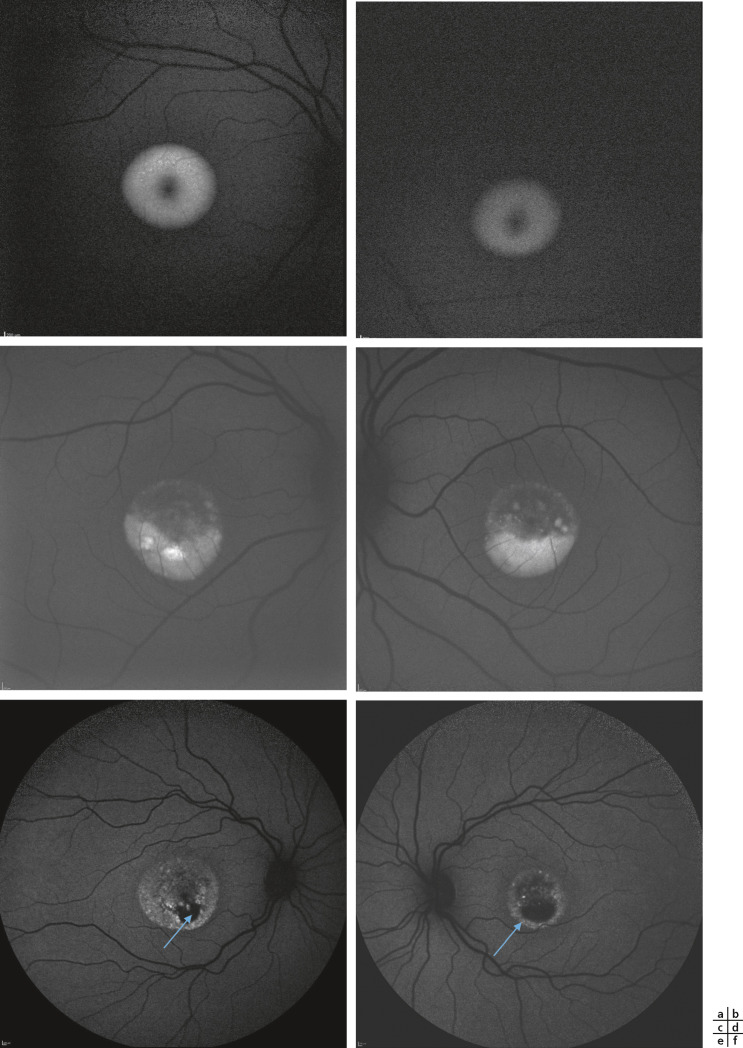
Fig. 17-12 Maladie de Best, imagerie en autofluorescence.
Au stade vitellin, le matériel est homogène et hyper-autofluorescent (a, b). Lors de la fragmentation, le matériel est déclive hyper-autofluorescent. À la partie supérieure de la lésion maculaire, les altérations de l’épithélium pigmentaire apparaissent sous forme d’alternance de petites lésions hypo- et hyper-autofluorescentes (c, d). L’atrophie géographique (flèches) est une évolution rare (e, f) et tardive.
Le diagnostic est simple en présence de matériel vitellin jaunâtre, hyper-autofluorescent, hyperréflectif en regard de l'épithélium pigmentaire, et d'un EOG altéré. Le diagnostic est parfois plus difficile dans les formes unilatérales ou compliquées de néovaisseaux ayant conduit à des lésions fibreuses ou pigmentées.
Un des diagnostics différentiels principaux est celui des maladies de Best récessives : les lésions sont multiples et l'ERG est altéré (fig. 17-14). Le pronostic visuel est plus sévère suite à des altérations marquées de l'épithélium pigmentaire ou à une fibrose maculaire.
Le pronostic visuel de cette dystrophie est relativement satisfaisant contrairement aux autres affections héréditaires : la plupart des patients gardent une acuité visuelle utile sur un des deux yeux (71 % des cas). Ces données sont à nuancer : 21 % des patients ont une acuité visuelle inférieure ou égale à 5/10 aux deux yeux (seuil autorisant la conduite automobile). L'enfant doit se surveiller œil par œil et consulter en urgence en cas de baisse d'acuité visuelle, de scotome dense ou de métamorphopsies afin de rechercher une complication néovasculaire.
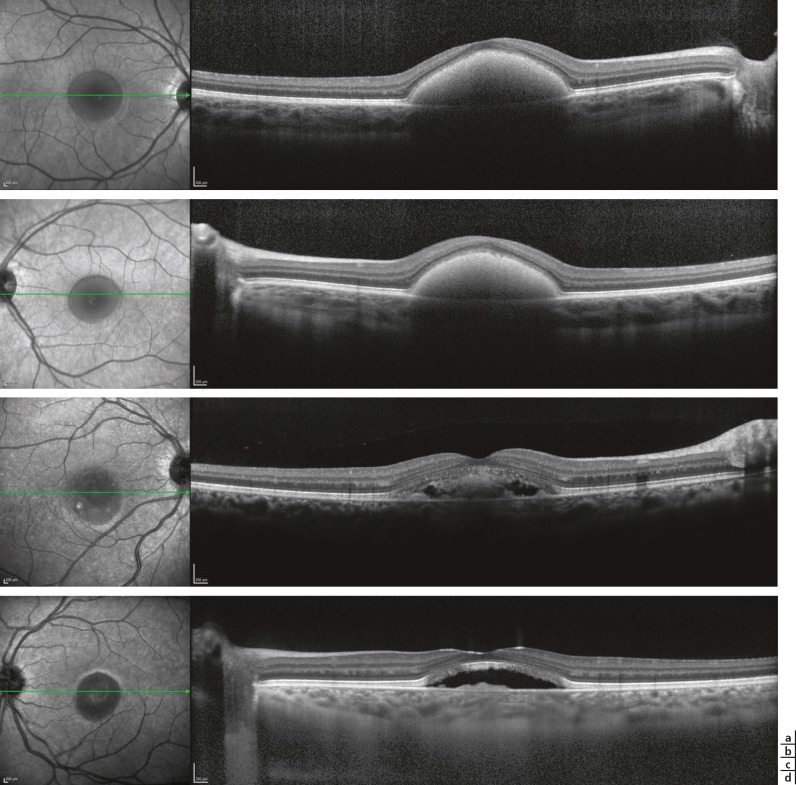
Fig. 17-13 Imagerie en SD-OCT, mode B.
Le matériel vitellin non fragmenté est hyperréflectif, il est situé sous la neurorétine et au-dessus de l’épithélium pigmentaire (a, b). Lors de sa fragmentation, le matériel est alors hyporéflectif reproduisant des aspects de faux décollement séreux rétinien (c, d).
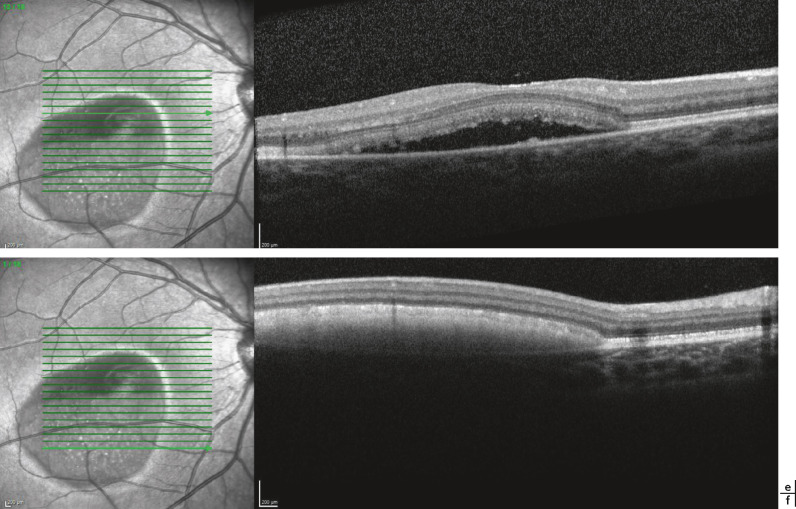
Fig. 17-13 Imagerie en SD-OCT, mode B. (Suite)
Les coupes e et f de l’oeil droit d’un même patient passent à la partie haute de la lésion sur une zone de matériel en voie de résorption hyporéflectif (e), et à la partie basse sur une zone de matériel vitellin hyperréflectif (f).
La maladie de Best a classiquement une transmission autosomique dominante mais la pénétrance et l'expressivité sont variables (porteur sain). Elle est liée à des mutations du gène BEST1 qui code pour une protéine transmembranaire de la membrane basolatérale de l'épithélium pigmentaire, la bestrophine [2, 3]. Cette protéine est impliquée dans le fonctionnement et le contrôle de canaux ioniques (canaux chlores calciumdépendants). Une forme récessive, rare et sévère, a été plus récemment décrite, avec des dépôts vitellins multiples disséminés à tout le pôle postérieur et une atteinte mixte de l'ERG (fig. 17-14). Il n'y a pas de traitement des dépôts vitellins. Les néovaisseaux choroïdiens surviennent dans 2 à 10 % des cas et peuvent être traités par injections intravitréennes d'anti-vascular endothelial growth factor (anti-VEGF) [8, 9].
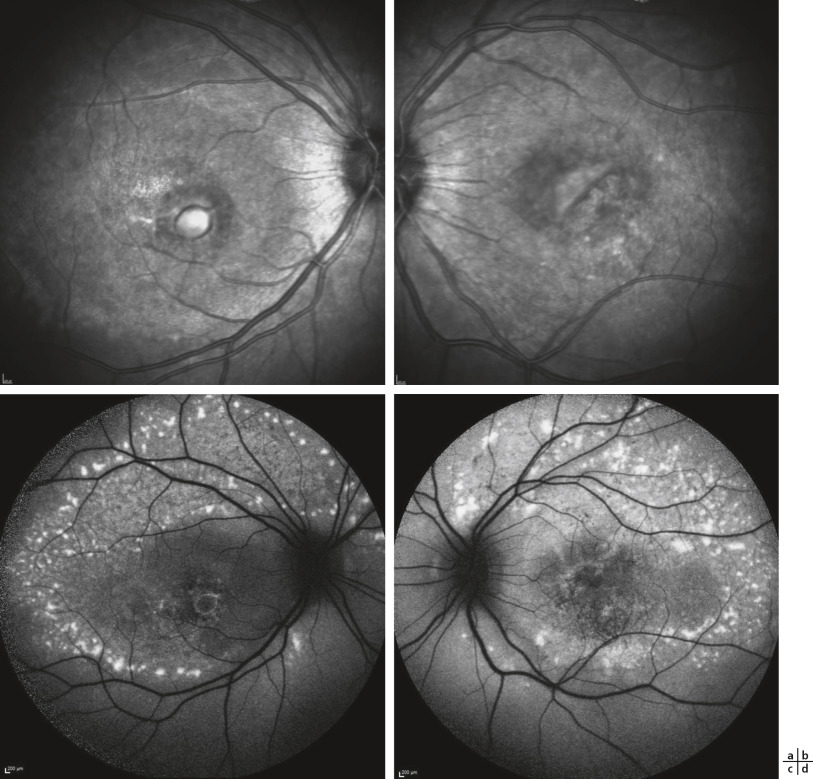
Fig. 17-14 Imagerie multimodale d’une forme récessive multifocale chez un enfant de 8 ans.
a, b. Infrarouge. c, d. Autofluorescence. Noter les dépôts vitellins multiples et disposés en couronne très à distance de la macula le long des arcades des vaisseaux temporaux. La macula est très remaniée avec des lésions de fibrose et un matériel hyporéflectif à l’oeil gauche.
[1] North V, Gelman R, Tsang SH Juvenile-onset macular degeneration and allied disorders Dev Ophthalmol: ( 2014 ) :53: 44-52
[2] Strauss RW, Ho A, Muñoz B The Natural history of the progression of atrophy secondary to Stargardt disease (ProgStar) studies Ophthalmology: ( 2016 ) :123: 817-828
[3] Song H, Rossi EA, Latchney L Cone and rod loss in Stargardt disease revealed by adaptive optics scanning light ophthalmoscopy JAMA Ophthalmol: ( 2015 ) :133: 1198-1203
[4] Zahid S, Jayasundera T, Rhoades W Clinical phenotypes and prognostic full-field electroretinographic findings in Stargardt disease Am J Ophthalmol: ( 2013 ) :155: 465-473 e3
[5] Boon CJF, Klevering BJ, den Hollander AI Clinical and genetic heterogeneity in multifocal vitelliform dystrophy Arch Ophthalmol: ( 2007 ) :125: 1100-1106
[6] Boon CJF, Theelen T, Hoefsloot EH Clinical and molecular genetic analysis of best vitelliform macular dystrophy Retina: ( 2009 ) :29: 835-847
[7] Meunier I, Manes G, Bocquet B Frequency and clinical pattern of vitelliform macular dystrophy caused by mutations of interphotoreceptor matrix IMPG1 and IMPG2 genes Ophthalmology: ( 2014 ) :121: 2406-2414
[8] Querques G, Bocco MCA, Soubrane G, Souied EH Intravitreal ranibizumab (Lucentis) for choroidal neovascularization associated with vitelliform macular dystrophy Acta Ophthalmol (Copenh): ( 2008 ) :86: 694-695
[9] Chhablani J, Jalali S Intravitreal bevacizumab for choroidal neovascularization secondary to Best vitelliform macular dystrophy in a 6-year-old child Eur J Ophthalmol: ( 2012 ) :22: 677-679
C. Hamel
Les rétinopathies pigmentaires sont des dégénérescences progressives des photorécepteurs (PR) de la rétine, évoluant sur de nombreuses années et aboutissant dans la majorité des cas à une cécité quasi totale. Le signe cardinal qui a donné son nom à cette maladie est la présence de dépôts pigmentaires tapissant le fond d'œil. Ils sont dÛs à des regroupements de cellules de l'épithélium pigmentaire de la rétine (EPR) autour des capillaires rétiniens, résultant de leur migration provoquée par la perte des PR. Nous décrirons d'abord les formes non syndromiques de rétinopathie pigmentaire qui correspondent à la classique rétinite pigmentaire, puis les rétinopathies pigmentaires syndromiques les plus fréquentes.
Fonctionnellement, la rétinite pigmentaire (RP) est d'abord marquée par une gêne visuelle voire une absence de vision en condition de faible éclairage dit scotopique, le soir (hespéranopie) et a fortiori la nuit (héméralopie), traduisant un dysfonctionnement des bâtonnets et contrastant avec une vision normale en condition d'éclairage diurne dit photopique. Cette déficience est la cause d'incidents qui restent assez peu fréquents dans le quotidien de l'enfant et n'attirent pas forcément l'attention s'il n'y a pas de contexte familial, la maladie restant ainsi ignorée. À ce stade aussi, l'examen du fond d'œil est peu informatif, il ne montre pas de dépôts pigmentaires mais seulement une rétine un peu terne en périphérie et un léger rétrécissement vasculaire rétinien (fig. 17-15a), requérant ainsi toute la perspicacité de l'ophtalmologiste pour faire les examens clés du diagnostic, c'est-à-dire l'OCT et l'ERG. C'est en général plus tard, à l'adolescence ou au stade d'adulte jeune, que s'installent progressivement des troubles du champ visuel périphérique en conditions diurnes, objectivés par les scotomes périphériques avec rétrécissement du champ visuel traduisant l'atteinte des cônes. À ce stade, les lésions classiques sont habituellement observables au fond d'œil telles que des pseudo-ostéoblastes, un amincissement vasculaire, une pâleur papillaire (fig. 17-15e). Finalement, vers 30 à 50 ans, les cônes fovéomaculaires finissent par disparaître lentement menant à une cécité totale. Ce tableau de la rétinite pigmentaire typique est dit à bâtonnets prédominants ou rod-cone dystrophy des Anglo-Saxons.
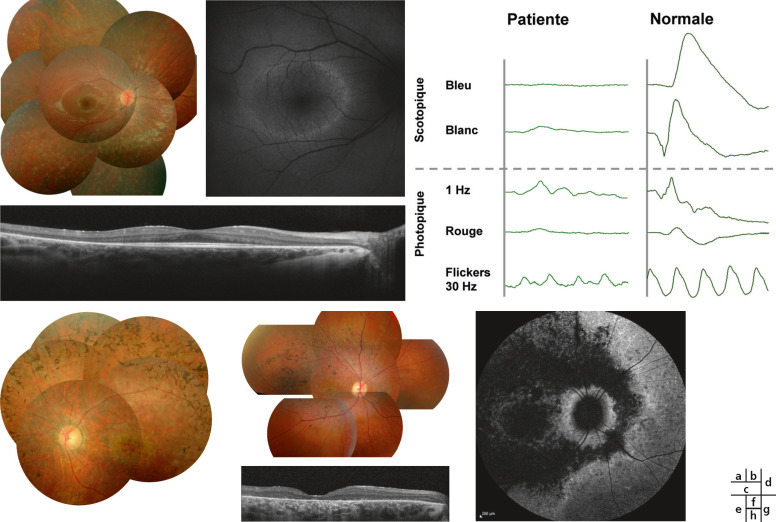
Fig. 17-15 Imagerie et électrophysiologie dans les rétinopathies pigmentaires.
a. Fond d’oeil droit d’une enfant de 12 ans avec une acuité visuelle de 10/10 montrant une macula normale avec la présence de petites taches blanches de dépigmentation en périphérie rétinienne et une arborescence vasculaire de calibre modérément diminué, sans pseudo-ostéoblaste. b. Son autofluorescence rétinienne révèle un anneau d’hyper-autofluorescence. c. OCT : un amincissement progressif de la zone ellipsoïde et de la couche nucléaire externe en dehors de la fovéa. d. L’ERG montre une quasi-absence de réponse en conditions scotopiques (bâtonnets), tandis que les réponses photopiques (cônes) sont hypovoltées mais encore bien enregistrables. e. Fond d’oeil gauche d’une fille de 12 ans ayant une forme juvénile sévère de rétinite pigmentaire liée au gène RDH12, montrant de nombreux pseudo-ostéoblastes et un rétrécissement important du réseau vasculaire rétinien. f. Fond d’oeil droit d’un garçon de 15 ans atteint d’une cone-rod dystrophy liée au gène ABCA4 causant habituellement la maladie de Stargardt. g. L’autofluorescence montre la perte importante d’autofluorescence maculaire avec une préservation péripapillaire. h. OCT : disparition de la zone ellipsoïde.
La mesure de l'acuité visuelle et le champ visuel, associant si possible une périmétrie cinétique en périphérie et statique sur les 30° centraux, sont évidemment indispensables pour évaluer les capacités fonctionnelles. Mais l'OCT est devenu essentiel, montrant l'amincissement des couches profondes de la rétine au-delà de la macula ou même en périphérie maculaire et contrastant avec une bonne conservation de ces couches en rétro- et périfovéolaire (fig. 17-15c). L'ERG, pratiqué avec des coques cornéosclérales, montre, même au stade débutant, des tracés scotopiques et photopiques nettement hypovoltés, avec néanmoins une prédominance de l'atteinte scotopique sur l'atteinte photopique (fig. 17-15d). Il deviendra rapidement éteint au fil des années, même chez des sujets ayant une acuité visuelle normale et manifestant peu de troubles du champ visuel dans la vie courante. L'ERG multifocal exige une bonne fixation pour être fiable, et n'a d'intérêt que si l'on veut étudier la fonctionnalité maculaire. Les PEV ainsi que l'EOG sont inutiles pour le diagnostic de RP. L'analyse de l'autofluorescence rétinienne qui remplace maintenant l'angiographie rétinienne montrera l'anneau d'hyper-autofluorescence maculaire caractéristique des RP et la présence de plages arrondies d'hypo-autofluorescence en périphérie, de taille et de nombre variables selon les sujets et le degré d'évolution (fig. 17-15b). Test de la vision des couleurs, sensibilité aux contrastes et adaptométrie sont des examens spécialisés qui ne sont intéressants qu'en cas de difficulté diagnostique particulière.
Appelée ainsi parce que la symptomatologie est initialement maculaire (baisse de l'acuité visuelle, photophobie, dyschromatopsie), relayée plus tard par une héméralopie souvent peu marquée et par des troubles du champ visuel périphérique, elle concerne moins de 10 % des patients. Dénommée aussi rétinite pigmentaire à cônes prédominants ou cone-rod dystrophy elle traduit une atteinte des cônes concomitante à celle des bâtonnets, voire prééminente. Elle cause un handicap plus sévère que dans la RP classique par la baisse d'acuité visuelle précoce. Les dépôts pigmentaires prédominent dans la région maculaire (fig. 17-15f) avec une autofluorescence maculaire diminuée (fig. 17-15g), un amincissement fovéolaire à l'OCT (fig. 17-15h) et un ERG où l'atteinte photopique est plus importante que l'atteinte scotopique.
Leur début est précoce, les premiers signes apparaissant vers l'âge de 2 à 3 ans. Elles évoluent rapidement, pouvant aboutir à la cécité à l'âge de 20 à 30 ans. La macula est en règle générale rapidement atteinte, entraînant baisse d'acuité visuelle, nystagmus et atrophie maculaire. L'ERG est éteint. Elles sont habituellement récessives autosomiques et ont des gènes en commun avec l'amaurose congénitale de Leber.
Il s'agit de cas qui peuvent passer inaperçus pendant toute l'enfance et l'adolescence, soit parce que la RP ne touche que le secteur rétinien inférieur, qu'elle est asymétrique ou unilatérale, soit parce que la pathogénicité est faible et l'évolution très lente, soit enfin par défaut de pénétrance du gène (porteurs asymptomatiques).
L'œdème maculaire est une complication de la RP, notamment dans les formes peu sévères et peut orienter à tort vers un diagnostic d'uvéite bilatérale ou de rétinoschisis. S'il englobe la papille, le diagnostic d'hypertension intracrânienne peut être porté par erreur. Dans tous les cas, il ne faut pas traiter par injection intravitréenne de corticoïdes mais utiliser les inhibiteurs de l'anhydrase carbonique. On peut voir aussi rarement un aspect de pseudo-Coats, toujours unilatéral. C'est une urgence nécessitant de traiter l'ischémie rétinienne.
Dans 55 % des cas, la maladie apparaît familiale et répond aux trois types possibles d'hérédité mendélienne. Les formes dominantes autosomiques sont les moins sévères alors que les formes liées à l'X, généralement associées à la myopie, le sont le plus. Dans 45 % des cas, la maladie apparaît sporadique, répondant le plus souvent alors à une hérédité récessive autosomique dans une petite fratrie ou plus rarement à une hérédité liée à l'X chez un garçon.
Plus de 70 gènes responsables sont actuellement recensés et seuls quelques-uns (RHO, RPGR, USH2A, EYS) concernent un nombre significatif de patients. Le séquençage simultané de ces gènes sera bientôt disponible en routine. Il est nécessaire pour un conseil génétique sÛr et pour inclure les patients dans des essais cliniques. Étant donné la complexité de la maladie, il est important que ces résultats génétiques soient donnés dans le cadre d'une consultation de génétique médicale ou dans un centre de référence/compétence pour les dystrophies rétiniennes.
Les héméralopies essentielles partagent avec la RP la cécité nocturne, mais elles sont congénitales et la vision reste stable au cours de la vie, sachant que dans certaines formes, souvent associées à la myopie, l'acuité visuelle est diminuée.
La choroïdérémie, dont la transmission est récessive liée à l'X, est une forme de rétinopathie pigmentaire difficile à diagnostiquer chez l'enfant, car l'atrophie choroïdienne est incomplète.
Les filles conductrices de choroïdérémie ou de RP liée à l'X ont une coloration hétérogène du fond d'œil. Ces altérations doivent être distinguées de la pathologie complète observée dans les cas masculins.
Enfin, toute destruction cellulaire sévère de la rétine d'origine inflammatoire, infectieuse ou toxique peut donner une pseudo-RP, mais le contexte doit orienter le diagnostic.
Classiquement, on propose le port de verres filtrants et la prise d'antioxydants. Chez l'enfant jeune sévèrement atteint, une aide scolaire est nécessaire. Chez l'enfant plus grand et l'adolescent, il faut rester très ouvert sur les possibilités professionnelles, tout en sachant que l'adolescent est souvent dans le déni, nécessitant une approche compréhensive mais ferme. Dans ce cadre, il est important d'expliquer l'essor des nouvelles thérapies (cellulaires, géniques et pharmacologiques).
Les rétinopathies pigmentaires syndromiques (RPS) les plus nombreuses forment le groupe des ciliopathies, car elles sont liées à des gènes impliqués dans la biologie du cil cellulaire, associant, à des degrés divers à la RP, des anomalies neurologiques, rénales, squelettiques et autres. La plupart de ces syndromes sont génétiquement hétérogènes. Nous les présentons par ordre de fréquence et par le signe associé le plus marquant, en indiquant l'hérédité autosomique dominante (AD) ou récessive (AR).
- – Surdité : lorsqu'elle est la seule association avec la RP, il s'agit du syndrome de Usher (11 gènes, AR) qui représente à lui seul 10 à 15 % de toutes les rétinopathies pigmentaires. C'est une surdité neurosensorielle congénitale profonde (type 1), sévère (type 2) ou rarement de début post-natal (type 3), nécessitant en règle générale une implantation cochléaire d'au moins une oreille avant l'âge de 1 an (type 1) ou un appareillage auditif (type 2). La rétinopathie est en tout point semblable à une RP typique, dont le diagnostic, souvent fait seulement à l'adolescence, est un choc psychologique chez un enfant déjà handicapé par la surdité.
- – Obésité et polydactylie : c'est le syndrome de Bardet-Biedl (19 gènes, AR), emblématique des ciliopathies, qui peut aussi associer une atteinte rénale, une lenteur d'idéation, un hypogénitalisme et des troubles endocriniens. La rétinopathie est habituellement sévère avec une atteinte maculaire précoce, souvent de type cone-rod dystrophy. Des mesures diététiques s'imposent avec un suivi endocrinien et une aide à la scolarité.
- – Ataxie : lorsqu'elle est cérébelleuse, il peut s'agir du syndrome de Joubert (10 gènes, AR), dont la rétinopathie peut être très sévère ou modérée, ou de l'ataxie spinocérébelleuse de type 7 (1 gène, AD) dont la cone-rod dystrophy est souvent inaugurale. Lorsqu'elle est de type neuropathie périphérique, il peut s'agir du syndrome NARP ou neuropathie, ataxie, rétinite pigmentaire (mutation T8993G de l'ADN mitochondrial).
- – Épilepsie, perte des acquis : chez un enfant de 5 à 10 ans, il faut redouter la lipofuscinose céroïde (5 gènes, AR) incluant la maladie de Batten pour le type 3, avec une rétinopathie à prédominance maculaire évoluant en quelques années vers une cécité totale et un pronostic vital sombre.
- – Néphronophtise : elle peut être associée au syndrome de Joubert ou bien former avec la RP, qui est habituellement précoce et sévère, le syndrome de Senior-Loken. Toute RP sévère de l'enfant doit ainsi faire pratiquer de principe un bilan rénal (échographie et bilan biologique).
- – Dysmorphisme facial : il doit évoquer le syndrome de Cohen (grandes incisives, jovialité, retard mental, neutropénie), les mucopolysaccharidoses (opacité cornéenne, hypertélorisme), le syndrome de Cockayne (aspect sénile), la maladie de Refsum infantile (visage allongé, atteintes sensorielles et neurologiques). À noter aussi la forme RP-microcéphalie avec lymphœdème inconstant.
- – Myopathie, ptosis, cardiopathie : ils doivent faire rechercher un syndrome de Kearn-Sayres ou d'autres atteintes mitochondriales, surtout en cas de surdité. Une cardiopathie doit faire rechercher un syndrome d'Alström. Notons encore les atteintes hépatiques (syndrome d'Alagille) ou les anomalies de l'émail dentaire (syndrome de Jalili).
C. Hamel
L'amaurose congénitale de Leber (ACL) est une forme de rétinopathie pigmentaire, particulièrement sévère, s'accompagnant de signes de malvoyance dès la naissance. Son principal diagnostic différentiel est l'achromatopsie congénitale.
À la naissance ou dans les semaines qui la suivent, les parents constatent chez leur enfant des mouvements erratiques des globes oculaires, sans contact oculaire et sans sourire réponse, avec fréquemment un signe digito-oculaire. L'examen révèle une absence de réaction à la lumière ou, plus fréquemment, une simple perception lumineuse avec une forte hypermétropie. Les segments antérieurs sont habituellement normaux et le fond d'œil n'objective pas de dépôts pigmentaires rétiniens. L'examen clé est l'ERG qui révèle l'absence de réponses à toutes les stimulations, tant photopiques que scotopiques. Par définition, il n'y a pas de signe extra-oculaire et l'imagerie par résonance magnétique (IRM) cérébrale n'est donc généralement pas nécessaire. Le diagnostic est confirmé par la découverte chez l'enfant d'une (homozygotie) ou deux (hétérozygotie composite) mutations parmi les 17 gènes responsables actuellement répertoriés, chacun des parents étant porteur hétérozygote permettant alors d'envisager des mesures préventives pour un autre enfant (diagnostic prénatal ou préimplantatoire). Il faut très rapidement mettre en œuvre un soutien à domicile (service d'accompagnement à l'acquisition de l'autonomie et à l'intégration scolaire [SAAAIS]) pour faciliter l'éveil et l'acquisition de la marche chez un enfant qui sera souvent très photophobe et expliquer la maladie aux parents qui pourront aussi bénéficier d'une aide psychologique. La maladie est classiquement peu évolutive.
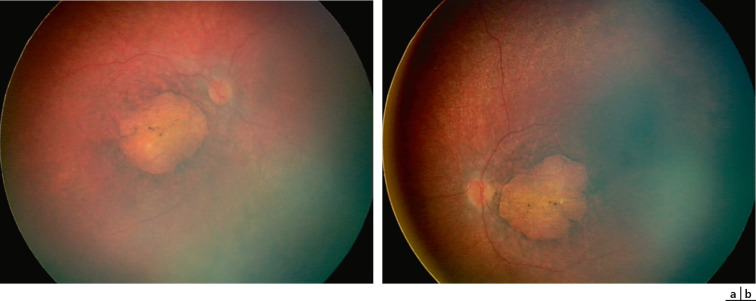
Fig. 17-16 a, b. Imagerie du FO (RetCam) d'un nourrisson de 3 mois sans éveil visuel présentant une forme d'amaurose congénitale « pseudo-colobomateuse maculaire » liée au gène NMNAT1 avec remaniement atrophique.
(Remerciements : Pr D. Denis.)
Les signes de malvoyance peuvent être moins marqués avec un nystagmus horizonto-rotatoire, l'absence d'hypermétropie, peu ou pas de photophobie mais au contraire une héméralopie et la constatation plus tard que l'enfant est capable de distinguer des objets ou des formes, de se diriger par lui-même, voire de lire avec une acuité visuelle très basse mais chiffrable. Ces formes moins dramatiques peuvent être dues à des mutations moins sévères des gènes d'ACL ou à des gènes particuliers (RPE65, LRAT). L'hérédité de l'ACL est toujours récessive autosomique sauf les très rares cas dominants dus à une mutation de novo de CRX L'association avec une cataracte ou un kératocône classiquement décrite est rare. Certaines formes génétiques particulières sont reconnaissables par des particularités du fond d'œil. Il peut s'agir d'un pseudo-colobome maculaire (NMNAT1) (fig. 17-16), de gros dépôts pigmentaires rétiniens (KCNJ13), d'une rétine épaissie avec préservation para-artériolaire de l'EPR (CRB1) ou encore de placards rétiniens blancs (NPHP5 et NPHP6). Ces derniers doivent faire rechercher une atteinte rénale et cérébelleuse (IRM cérébrale), car ils peuvent aussi être responsables des syndromes de Joubert ou de Senior-Loken.
Outre l'ACL, l'existence d'une malvision congénitale doit aussi évoquer un retard de maturation des voies visuelles, une forme sévère de RPS (syndromes de Joubert, de Senior-Loken), une maladie métabolique, une encéphalopathie, une embryofœtopathie ou, pour les formes moins sévères, la cécité nocturne congénitale stationnaire et l'albinisme. En général, l'existence de signes associés oriente vers ces diagnostics différentiels. Reste l'achromatopsie congénitale qui est trompeuse car sa présentation ressemble à celle de l'ACL. Il s'agit d'une pathologie oculaire isolée avec, dès la naissance, un nystagmus, une hypermétropie fréquente et une acuité visuelle basse. La photophobie qui se développe rapidement est majeure, au point que l'enfant paraît aveugle en conditions diurnes alors qu'il pourra lire et se repérer dans la pénombre. L'ERG est éteint en conditions photopiques mais, à la différence de l'ACL, les réponses sont bien enregistrables en conditions scotopiques montrant la nécessité de pratiquer cet examen dans les règles, même chez le bébé ou le jeune enfant. La vision des couleurs est nulle ou très partielle. La maladie est récessive autosomique et est due à des mutations de certains gènes s'exprimant seulement dans les cônes. L'OCT montre d'ailleurs un amincissement voire une lacune fovéolaire. Il existe une forme liée à l'X présente uniquement chez les sujets masculins, qui correspond précisément au diagnostic de monochromatie à cônes bleus, due à des mutations des opsines rouge/vert. À la différence de l'ACL, les achromates ont un champ visuel périphérique utile en condition mésopique ou scotopique et conservent toute leur vie une capacité de lecture avec des dispositifs grossissants. Leur parcours scolaire peut s'effectuer dans des classes normales.
G. Le Meur, C. Hamel
La recherche génétique et physiopathologique a ouvert un champ immense aux explorations de stratégies thérapeutiques nouvelles. De nombreuses connaissances ont été acquises sur les causes génétiques de ces maladies avec actuellement plus de 250 gènes répertoriés responsables de dystrophies de la rétine, agissant dans de nombreux compartiments des cellules rétiniennes (surtout photorécepteurs mais aussi épithélium pigmentaire de la rétine), aussi divers que la transduction visuelle, la machinerie ciliaire, la transcription et l'épissage, le métabolisme énergétique, etc. Toutefois, les voies métaboliques aboutissant à la mort des cellules restent encore mal définies. Les traitements ciblent donc les causes primaires des pathologies, à savoir les gènes, mais visent aussi à ralentir le processus dégénératif, à réparer les tissus par apport de cellules nouvelles, voire à restaurer la vision avec des prothèses de vision artificielle. La thérapie génique amène, dans les cas de pertes de fonctions, l'acide désoxyribonucléique complémentaire (ADNc) normal du gène muté. Cette approche a déjà prouvé son efficacité dans une forme particulière d'amaurose congénitale de Leber liée au gène RPE65 Fort de ces premiers résultats, d'autres essais ont démarré pour la maladie de Stargardt, le syndrome de Usher, la neuropathie optique héréditaire de Leber, l'achromatopsie congénitale, la choroïdérémie. Plusieurs années seront nécessaires pour établir l'efficacité de ce traitement car l'évolution naturelle de ces maladies est lente. Des dispositifs artificiels visant à remplacer la mosaïque des photorécepteurs ou à stimuler les cellules ganglionnaires de la rétine ont déjà fait l'objet d'essais cliniques. Les résultats sont spectaculaires, des patients préalablement aveugles redevenant capables de lire de gros caractères. La thérapie cellulaire est aussi à l'étude, mais la stabilité phénotypique et le grand nombre de cellules à implanter sur une petite surface posent encore des problèmes. Enfin, la récente découverte que l'on peut rendre photosensible des cellules de la rétine non sensorielles selon une technologie dénommée optogénétique ouvre des perspectives d'application. Ces importantes évolutions technologiques ont ainsi beaucoup modifié notre relation aux patients qui réalisent que recouvrer la vision n'est plus une utopie.