Les bases
Aspects fondamentaux
Coordonné Par D. Denis, A. Aziz-Alessi
S. Creuzet, H. Etchevers
Afin d'appréhender les mécanismes qui sous-tendent la physiologie de la vision, la connaissance des bases embryologiques du développement de l'œil et de ses annexes est un prérequis indispensable. La morphogenèse oculaire débute au cours de la 4 semaine de vie embryonnaire, alors que l'ébauche oculaire s'individualise des diverticules latéraux du cerveau antérieur par des mouvements morphogénétiques complexes. Elle sollicite la contribution respective des divers feuillets de l'embryon, le neuro-ectoderme, l'ectoderme de surface, le mésoderme et les cellules de la crête neurale, pour l'élaboration de ses différentes composantes. Les perturbations des interactions cellulaires et des mécanismes moléculaires mobilisés au cours de ces étapes critiques sont responsables d'anomalies congénitales variées. Nous évoquerons, à cet égard, les processus embryologiques dont les dérégulations sont à l'origine des malformations oculaires et qui font, plus particulièrement, l'objet de chapitres détaillés dans cet ouvrage.
Au stade précoce du développement, dès 2 semaines après la fécondation, l'embryon est composé de deux feuillets cellulaires superposés : l'un, sus-jacent, l’épiblaste ou ectoderme primitif, et l'autre, sous-jacent, l’hypoblaste ou endoderme primitif. Formés de cellules cohésives, ces feuillets épithéliaux constituent, dans leur zone de contact, le disque embryonnaire à partir duquel se développe l'embryon. Au-delà, les feuillets se poursuivent séparément et délimitent les vésicules extra-embryonnaires, destinées à former les annexes extra-embryonnaires, à savoir l'amnios et le placenta, situés respectivement au-dessus et en dessous du disque embryonnaire.
Orientation des axes embryonnaires
L’organisation spatiale et la dynamique des mouvements morphogénétiques impliquent de fixer des axes de référence anatomique internes à l’embryon. L’embryon humain présente l’avantage – par rapport à certains organismes modèles en embryologie tels que la souris ou le poisson – de se développer dans le même plan de l’espace, au cours des périodes critiques conduisant de la gastrulation à la neurulation. Alors que l’embryon est encore diblastique, c’est-à-dire composé de deux feuillets, un plan d’organisation « dorsoventrale » peut lui être assigné. L’acquisition de cette polarité repose sur le fait que l’épiblaste, à l’origine du système nerveux central, se développe au pôle dorsal de celui-ci alors que l’hypoblaste, destiné à tapisser la face interne de cavités viscérales, détermine le pôle ventral. Lorsque la gastrulation s’engage, l’invagination des cellules d’épiblaste en position intermédiaire conduit à la formation d’un troisième feuillet embryonnaire, le mésoderme. Cette organisation triblastique mobilise les cellules de l’épiblaste qui convergent vers une ligne longitudinale médiane, la ligne primitive où elles subissent un double changement : d’une part, elles cessent d’être ectodermiques pour s’intercaler entre l’épiblaste et l’hypoblaste, et deviennent mésodermiques ; d’autre part, elles adoptent une organisation tissulaire moins dense et cohésive que celle du feuillet épithélial, le mésenchyme, propice à des remodelages tissulaires rapides et des migrations à distance. La formation de la ligne primitive confère le plan de symétrie bilatérale qui sépare les côtés droit et gauche de l’embryon, et donne également les repères d’un axe « médiolatéral ». Chez tous les organismes bilatériens, la morphogenèse de l’embryon débute au pôle céphalique, qui définit la partie antérieure ou rostrale de celui-ci, puis gagne de proche en proche les niveaux plus postérieurs ou caudaux. Par conséquent, ce gradient de développement permet de définir précocement l’axe « antéropostérieur » ou « rostrocaudal » de l’embryon.
Dans les jours qui suivent, un sillon médian se forme à la surface de l'épiblaste, dont la position définit l'extrémité caudale de l'embryon. Ce sillon s'appelle la ligne primitive et marque l'endroit où se déroule la gastrulation (fig. 28-1 a et b). Pour reprendre l'aphorisme du grand embryologiste Lewis Wolpert, « le moment le plus important de la vie n'est ni la naissance, ni le mariage, ni la mort, mais la gastrulation » [1]. À partir de ce stade, l'épiblaste du disque embryonnaire proprement dit devient l'ectoderme définitif à l'origine du tissu épithélial qui tapisse et couvre la face externe de l'organisme. Lorsque la ligne primitive est à son extension maximale, les cellules les plus rostrales situées à sa base s'invaginent et s'agrègent en un amas de cellules mésodermiques axiales (fig. 28-1b et c). La ligne primitive engage ensuite une régression rostrocaudale, relative par rapport à l'allongement antérieur de l'embryon, au cours de laquelle elle dépose dans son sillage le matériel cellulaire destiné à former la notochorde. De façon concomitante, la plaque neurale est induite dans l'ectoderme médial sus-jacent. Bien qu'initialement formée d'une seule couche de cellules, la plaque neurale se caractérise par un rapide épaississement, qui conduit à la spécification du neuro-ectoderme et à sa démarcation de l'ectoderme latéral. La plaque neurale subit une réorganisation cellulaire et des mouvements complexes d'extension et de convergence qui précédent la formation d'une gouttière neurale (fig. 28-1 d). Les bords latéraux de cette gouttière se rejoignent progressivement pour fusionner le long de la ligne médiane dorsale. La fusion des bords du neuro-ectoderme permet d'une part, de restaurer la continuité de l'ectoderme superficiel, destiné à former l'épiderme, et d'autre part, d'internaliser le tube neural à l'origine de l'ensemble du système nerveux central (fig. 28-1e).
La fermeture du tube neural débute au niveau du futur cerveau moyen, puis gagne, de façon bidirectionnelle, les niveaux plus rostraux et plus caudaux. En amont, le mécanisme laisse un neuropore antérieur qui se résorbe dans les jours qui suivent (fig. 28-1 c et h). Les anomalies du développement qui surviennent au cours de ce processus de fermeture génèrent des malformations extrêmement sévères telles que l'anencéphalie et qui ne sont pas compatibles avec la vie postnatale. Celles-ci peuvent être facilement et précocement décelées par échographie. La désorganisation extrême qui en résulte dans le tissu cérébral a pour conséquence des remodelages importants et délétères des champs optiques, du fait de leur proximité.
Outre l'implication successive de l'ectoderme, du neuro-ectoderme et, dans une moindre mesure, du mésoderme, l'ontogenèse de l'œil mobilise une ultime population cellulaire qui contribue de façon essentielle à la morphogenèse, l'organogenèse et la physiologie optique : la crête neurale. Il s'agit d'une population de cellules qui a pour origine les bourrelets neuraux qui délimitent latéralement la gouttière neurale (fig. 28-2 a). Avant la fermeture du tube, ces cellules sont épithéliales et liées au neuro-ectoderme, mais, à mesure que la fermeture du tube neural s'engage, elles se détachent des bourrelets latéraux et deviennent mésenchymateuses (fig. 28-2 a et b). Leur individualisation s'opère selon une cinétique bidirectionnelle qui suit la fermeture du tube neural. Bien que ce processus soit très conservé chez les Vertébrés, des variations subtiles peuvent exister dans la dynamique de leur délamination selon les espèces.
La crête neurale est une grande innovation qui a marqué l'histoire des Chordés et constitue une caractéristique exclusive des Vertébrés. Du fait de son caractère hautement multipotent, elle est considérée comme le quatrième feuillet germinatif de ce groupe phylogénétique. Son apparition au cours de l'évolution a permis l'acquisition d'une grande variété de caractères propres, parmi lesquels la formation d'un squelette craniofacial comprenant les mâchoires, la face supérieure et le crâne. En outre, l'émergence de ces structures squelettiques a coïncidé avec l'accroissement et la sophistication du cerveau antérieur et des organes des sens.
Il est plus approprié de parler de « cellules de la crête neurale » que de « crêtes neurales », qui désignent spécifiquement les bords de la gouttière neurale en cours de fermeture. Les cellules de la crête neurale (CCN), lorsqu'elles se détachent des bourrelets neuraux, démarrent d'importantes migrations qui les conduisent à essaimer dans tout l'embryon où elles se différencient en une remarquable variété de lignages et de dérivés [2]. Les dérivés des CCN des niveaux céphaliques et troncaux sont présentés dans la figure 28-2c .
Outre une contribution particulièrement riche à l'ontogenèse, la crête neurale subsiste également chez l'adulte à l'état indifférencié, au niveau céphalique, dans certains foyers qui se comportent comme autant de réservoirs ou « niches » de cellules souches, susceptibles de participer à des processus régénératifs variés [3]. Du fait de leurs capacités de différenciation plus étendues par rapport à celles du mésoderme, les cellules souches de la crête neurale font l'objet d'intenses recherches visant à maîtriser les conditions de leur utilisation pour l'ingénierie tissulaire et la médecine régénérative. Ceci est particulièrement le cas en ce qui concerne la cornée.

Fig. 28-1 Vue dorsale en stéréomicroscopie d'un embryon humain au jour gestationnel 18 (J18) après fécondation. La partie caudale, en bas, est toujours en cours de gastrulation pendant que la partie rostrale entame déjà la neurulation. Vue dorsale d'un embryon du même stade par microscopie électronique à balayage (zone agrandie en c). À l'extrémité la plus rostrale de la ligne primitive, des cellules de l'épiblaste se détachent et migrent en tant que mésenchyme lâche dans le sens des flèches entre épiblaste et hypoblaste mais aussi vers la tête, déposant progressivement tout le mésoderme suivant une distribution rostrocaudale. J21. La gouttière neurale est ouverte dans la région céphalique, vers le haut. Vue dorsale, tête à gauche, J21. Le mésoderme s'organise en paires de blocs épithéliales, les somites, de part et d'autre de la gouttière neurale, à l'origine des futures structures segmentées du corps, à savoir vertèbres, côtes et muscles. Au début de la 4 semaine de gestation, le tube neural se ferme et se détache sous l'ectoderme dorsal, alors que les somites continuent à se former en l'accompagnant de rostral en caudal. En vue frontale de la face présomptive, la fermeture du tube neural n'est pas encore achevée, laissant apercevoir le prosencéphale. L'ébauche du cœur se développe à proximité du cerveau antérieur avant de s'en éloigner par la formation des arcs pharyngés au cours de la semaine qui suit. La partie restant ouverte du tube neural en rostral est le neuropore antérieur, J26. Le neuropore postérieur est encore ouvert au cours de la 4 semaine de gestation. Ant : antérieur; Di : diencéphale; Mes : mésencéphale; Post : postérieur; Pros : prosencéphale; Rh : rhombencéphale; So : somite. (Remerciements au Dr K.-K. Sulik. Reproduction autorisée.)
Le développement de l'œil proprement dit débute à 22 jours de gestation (J22), alors que la taille de l'embryon humain atteint 2 mm de longueur. Au niveau céphalique, tandis que la plaque neurale commence à se replier pour former le tube neural, des dépressions ou diverticules apparaissent à la face interne de la plaque et marquent des évaginations latérales du neuro-ectoderme vers l'ectoderme de surface (fig. 28-3 a, c et d). À ce niveau, la partie médiale de la plaque neurale est destinée à former une division majeure du cerveau antérieur, le diencéphale, à partir duquel se forment d'autres structures telles que l'hypothalamus et le chiasma des nerfs optiques, pour une distribution des axones indispensable à la vision binoculaire. Une seconde division majeure se forme dans la partie latérale de la plaque neurale antérieure : il s'agit du télencéphale, à l'origine des hémisphères cérébraux qui vont croître en avant des diverticules optiques (fig. 28-3 b et e).
Dans les jours qui suivent, alors que des unités métamériques de mésoderme troncal, les subissent une ségrégation de part et d'autre du tube neural en suivant l'élongation du corps (fig. 28-1 f et fig. 28-1g), les vésicules optiques issues des évaginations du neuro-ectoderme s'élargissent (fig. 28-2 a, b et fig. 28-3e). La progression des vésicules optiques s'opère en direction de l'ectoderme de surface au contact duquel le neuro-ectoderme s'épaissit et détermine le disque rétinien vers J27. Leur croissance latérale est accompagnée par un afflux de cellules mésenchymateuses (fig. 28-4 ).
De façon réciproque, l'ectoderme de surface subit également une différenciation qui débute, là encore, par l'épaississement des cellules à son niveau (fig. 28-4 b). Cet épaississement délimite la placode cristallinienne qui secondairement s'invagine jusqu'à former une vésicule cristallinienne (fig. 28-4 b et c), puis s'individualise totalement de l'ectoderme de surface adjacent pour aboutir à la formation d'une lentille internalisée sous l'ectoderme, le cristallin. La formation de la placode cristallinienne est l'un des exemples les plus classiques du processus d'induction en biologie du développement, mettant en jeu des signaux morphogénétiques produits par le neuro-ectoderme et l'ectoderme de surface, et sollicitant également d'autres tissus, situés plus à distance tels que le mésoderme cardiaque et l'endoderme pharyngien.
La formation de la placode cristallinienne coïncide avec l'apparition d'une constriction à la face proximale de la vésicule optique, au niveau de son point d'attache à la paroi latérale du cerveau antérieur. Cette constriction, la tige optique, s'allonge et s'accentue au cours de la croissance à mesure que la morphogenèse de la vésicule optique gagne en sophistication. La lumière de la tige optique maintient une continuité entre la cavité de la vésicule optique, qui donne l'espace sous-rétinien, et le troisième ventricule, vésicule unique et médiale du diencéphale (fig. 28-4 c).
À la fin de la 4 semaine de développement, la vésicule optique est globalement sphéroïde et composée d'une monocouche de cellules. Elle subit ensuite une invagination spectaculaire par le biais d'élongations et de mitoses cellulaires qui accroissent la surface de tissu neuro-épithélial à son niveau, mais également par des changements cytosquelettiques et des phénomènes de repliement qui aboutissent à la formation de la cupule optique.
Le disque rétinien, situé initialement à l'apex de la vésicule (fig. 28-4 c), est transitoirement superposé à la placode du cristallin : ces deux couches cellulaires d'origine distincte sont liées par des pontages cellulaires temporaires. L'accroissement de la cupule optique n'étant pas uniforme à sa circonférence, une croissance différentielle conduit à la formation d'un sillon le long de la face distale et ventrale, dont les bords convergent pour former la fissure optique. À J29, deux invaginations concomitantes – du disque de la rétine et de la placode du cristallin – sont presque achevées (fig. 28-4 d). Superficiellement, une petite dépression peut être observée alors que la lentille du cristallin est en cours d'internalisation. Autour de ce point, le territoire où l'ectoderme de surface tend à recouvrer son intégrité est à l'origine de la future cornée.
La vésicule du cristallin se sépare définitivement de l'ectoderme de surface avant J36. Les cellules épithéliales du cristallin se referment sur une cavité et sont bordées extérieurement par une lame basale qui forme la capsule du cristallin. Au niveau de la fissure optique, le sillon longitudinal s'étend de la tige optique jusqu'à la cupule qui, parallèlement, s'élargit et s'invagine. Ce mouvement morphogénétique aboutit à la juxtaposition de la paroi distale et de la paroi proximale de la tige optique. Dans la fissure, une branche de l'artère ophtalmique, l'artère hyaloïde et des cellules dérivées de la crête neurale se trouvent incorporées à l'espace lentorétinal. À la fin de la 6 semaine de développement (6 sd), soit approximativement à 8 semaines d'aménorrhée, les bords de la fissure se rejoignent et fusionnent en isolant dans le centre de la tige optique les vaisseaux hyaloïdes et le mésenchyme associé, à l'origine de l'artère et de la veine centrale de la rétine (fig. 28-5 a). La fermeture de la fissure optique commence au milieu de la tige optique et continue simultanément dans une direction proximale (vers le cerveau) et distale (vers la rétine). La fusion de la fissure s'achève en marge de la cupule optique en ménageant un orifice à l'origine de la pupille (fig. 28-5 b).
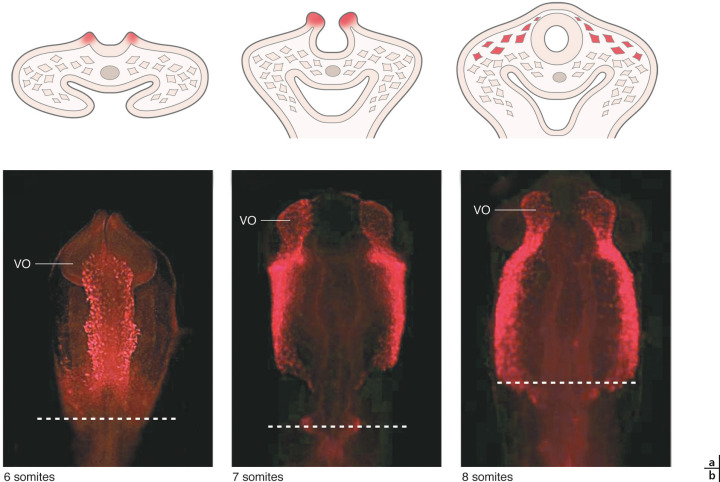
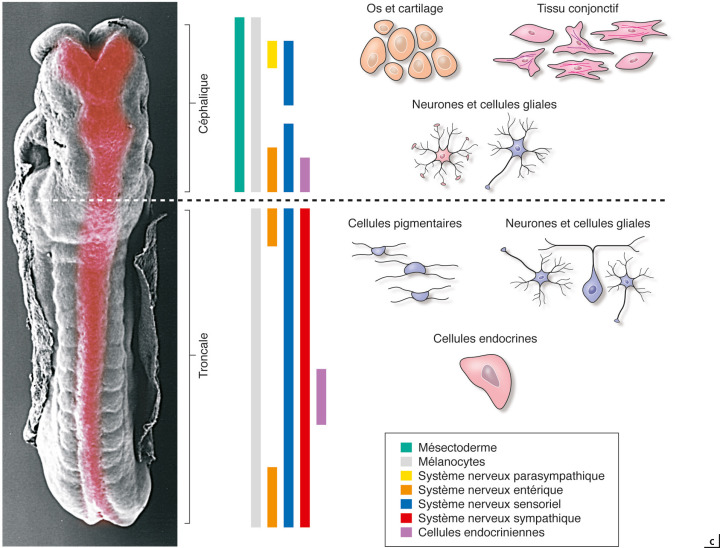
Fig. 28-2 Les cellules de la crête neurale (CCN), lorsqu’elles se détachent des bourrelets neuraux, débutent d’importantes migrations qui les conduisent à essaimer dans tout l’embryon.
a. Schémas en vue transversale au niveau céphalique : CCN (rouges) initialement au sein des bourrelets neuraux, puis en tant que mésenchyme qui migre à distance du tube neural. b. Photomicrographies en vue dorsale de la région céphalique d’embryons de poulet autour de 30–35 h d’incubation, avec 6 à 8 paires de somites et rostral vers le haut ; niveau de coupe indiqué par un trait. Les CCN sont marquées en fluorescence rouge. Elles se détachent du tube neural dorsal en même temps que les vésicules optiques (VO) s’évaginent progressivement du diencéphale latéral, sous cette chape de mésenchyme.c. Embryon humain, vue dorsale en microscopie électronique à balayage, vers le 24e jour de gestation. Tube neural indiqué en rouge. Les dérivés cellulaires des CCN proviennent de différents niveaux le long du tube neural, et sont plus nombreux à partir des CCN céphaliques que troncales. Certains dérivés, tels les tissus structuraux issus du mésectoderme comme la sclère ou les os de la face, sont produits uniquement par des CCN céphaliques ; d’autres, tels certains dérivés endocriniens ou le système nerveux entérique, ne proviennent que de régions très spécifi ques, délimitées selon le niveau de l’axe rostrocaudal. (fig. 28-2 c : remerciements au Dr K.-K. Sulik. Reproduction autorisée.)
Colobome
Le terme « colobome » signifie « mutilation » en grec. Cependant, dans la pratique clinique, il désigne un défaut congénital du quadrant inféronasal de l’oeil, intéressant l’iris, l’uvée et la rétine. Les colobomes sont généralement sporadiques et bilatéraux. Ils sont le résultat d’une absence totale ou partielle de la fermeture de la fissure optique qui conduit aux phénotypes rencontrés. Ainsi, les colobomes peuvent être antérieurs, postérieurs (rétine, choroïde, nerf optique) ou antéropostérieurs pour les formes les plus graves. Cette malformation a pour conséquence un défaut de l’induction et de la formation des tissus de l’uvée.
Le colobome de l’iris apparaît comme étant un défaut inféronasal affectant le stroma, le muscle lisse et l’épithélium pigmentaire à ce niveau.
Le colobome de l’uvée se caractérise par l’absence de procès ciliaires et une atrophie du muscle ciliaire. Ces structures, en condition physiologique, garantissent l’intégrité de la chambre antérieure de l’oeil en assurant deux fonctions essentielles : d’une part mécanique, grâce aux fibres zonulaires qui permettent le maintien du cristallin par des ligaments suspenseurs des corps ciliaires, et d’autre part physiologique, puisque les procès ciliaires sécrètent l’humeur aqueuse. Dans un contexte colobomateux, le cristallin adjacent est en retrait, en raison d’une hypoplasie ou d’une insuffisance des fibres zonulaires. Dans les colobomes syndromiques associés à des malformations complexes (telles que certaines trisomies), une différenciation anormale du mésenchyme dans l’espace rétrocristallinien peut survenir avec la formation ectopique d’autres dérivés de CCN, tels que du tissu adipeux ou du cartilage (fig. 28-5 c).
Les colobomes choriorétiniens impliquant la rétine peuvent présenter d’importants déficits pour la fonction visuelle. À proximité du colobome, la prolifération du tissu neuroblastique rétinien peut conduire à la formation de rosettes. Un défaut d’induction ou de différenciation de l’épithélium rétinien pigmentaire dans la zone du colobome est souvent associé à l’absence de la membrane de Bruch et du tissu choroïdien, alors que la sclérotique sous-jacente paraît normale. Ces colobomes postérieurs et inféronasaux sont ceux rencontrés dans l’association syndromique connue sous l’acronyme CHARGE pour Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital anomalies, Ear anomalies (colobome, maladies cardiaques, atrésie des choanes, retard de croissance et/ou de développement psychomoteur ; hypoplasie génitale, malformation de l’oreille), et laissant suspecter des mutations du gène CHD7.
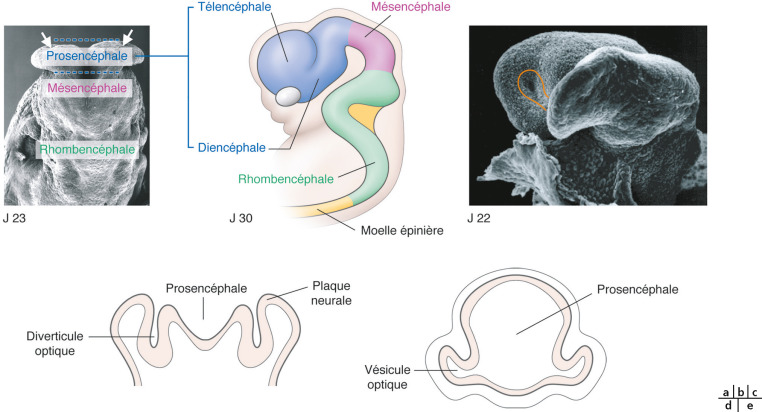
Fig. 28-3 Les régions du cerveau se différencient avant la fin du premier mois de grossesse.
a. Les évaginations optiques (flèches) sont visibles dans le neuro-épithelium dès le 22e jour (J) de gestation. b. Les sous-divisions anatomiques du cerveau sont indiquées ; vue latérale gauche. La vésicule optique gauche figure en blanc. c. Vue frontolatérale d’un embryon humain à J22 en microscopie électronique à balayage ; le diverticule optique est délimité en orange. d, e. Schémas en coupe transversale des changements morphogénétiques du cerveau antérieur et champs optiques entre les 22e (d) et 26e (e) jours de gestation et la fermeture des bourrelets du diencéphale. (fig. 28-3 a et c adaptées de Dr K.-K. Sulik, avec autorisation ; fig. 28-3 b adaptée de Wikimedia Commons ; fig. 28-3 d et e adaptées de Larsen WJ. Essentials of human embryology, avec l’accord d’Elsevier.)

Fig. 28-4 Induction du cristallin et morphogenèse de la cupule optique.
a. À la fin du 1er mois de gestation, plusieurs mouvements tissulaires se passent simultanément dans la vésicule optique (flèche). b. Ces illustrations de coupes histologiques chez deux embryons humains du même stade montrent la fugacité des étapes qui marquent le passage de l’induction du placode cristallinien et du disque rétinienne à la démarcation de la tige optique et l’oblitération de l’espace sous-rétinienne (ESR). c. La cupule et la tige optique proviennent du neuro-ectoderme. Sous l’ectoderme, des cellules mésenchymateuses (M), essentiellement d’origine crête neurale, entourent la cupule optique. d. Vue en coupe partielle montrant la fissure optique sur la face inférieure de la tige, qui résulte du contact de la rétine neurosensorielle avec la couche du futur épithélium pigmentaire rétinien (EPR). Une animation de ce processus peut être visionnée à l’adresse Internet suivante : http://www.nature.com/nrn/journal/v8/n12/extref/ nrn2283-s1.swf. (fig. 28-4 b et c : adapté de [27].)
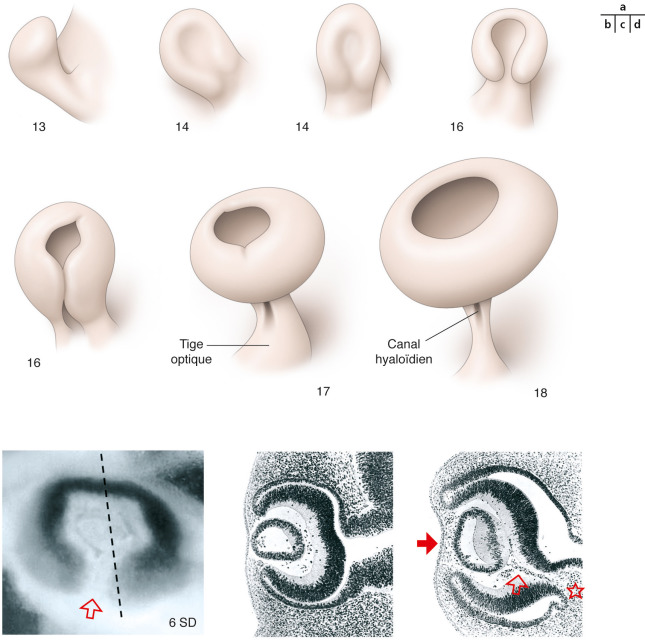
Fig. 28-5 Fermeture de la tige optique et développement des vaisseaux hyaloïdes à la fin de la 6e semaine du développement (SD).
a. Entre les stades 13 et 18 de Carnegie, c’est-à-dire entre 5 et 8 SD chez l’embryon humain, plusieurs mouvements morphogénétiques se coordonnent dans la région optique pendant la transformation de la vésicule à une structure oculaire plus complexe. b. OEil microdisséqué d’embryon humain vers J37, montrant la fissure optique (flèche) côté ventral et la pigmentation de l’épithélium pigmentaire rétinien par transparence. c. Coupe histologique dans le plan délimité en (b). La constriction de la tige optique et de l’espace sous-rétinien est évidente, ainsi que le détachement de la vésicule cristallinienne. d. Coupe histologique dans le même plan, vers J42. Des cellules mésenchymateuses se trouvent entre l’ectoderme et le cristallin (flèche pleine), ainsi qu’en accompagnement (étoile) des vaisseaux hyaloïdes (flèche vide) dans la fissure optique. (fig. 28-5 a : adapté de [27]. Fig. 28-5b-d : images des auteurs.)
Après la séparation de la vésicule cristallinienne de l'ectoderme de surface, ce dernier se referme pour donner le futur épithélium cornéen (fig. 28-6 a). De façon concomitante, à J39, une vague de cellules mésenchymateuses d'origine des CCN migre massivement le long de la cupule optique et directement sous l'ectoderme de surface (fig. 28-6 c à e). Cette migration s'opère selon trois vagues successives. Les premières cellules à coloniser ce territoire s'accumulent à proximité du cristallin : elles adoptent une morphologie pavimenteuse et développent des contacts apicolatéraux. Ces contacts organisent des jonctions intercellulaires continues qui aboutissent à la formation de l'endothélium cornéen et du trabéculum. Deux vagues successives de mésenchyme viennent secondairement élaborer d'abord le stroma de la cornée, ensuite le stroma de l'iris et le mésenchyme de l'angle iridocornéen (fig. 28-6 d). À noter que l'épithélium cornéen reste d'origine ectodermique (fig. 28-6 e) [4,5].
À la fin de la période embryonnaire (à la fin de 8 sd), la rétine est clairement structurée en deux composantes majeures étroitement contiguës. Extérieurement, la couche mince de la cupule optique forme l'épithélium pigmentaire rétinien (EPR); elle est doublée intérieurement d'une couche tissulaire beaucoup plus épaisse, destinée à former la rétine neurale (fig. 28-5 b et d). Ces deux couches sont séparées par un espace sous-rétinien étroit, vestige de la cavité ventriculaire de la vésicule optique. Vers 5 sd, l'accumulation de la mélanine peut être déjà mise en évidence dans l'EPR. La rétine neurale débute une différenciation centrifuge à partir de la couche intérieure neuroblastique située près de la tige optique. Simultanément, la cavité du cristallin disparaît par l'allongement considérable des cellules postérieures qui sont disposées parallèlement et organisent ainsi les fibres primaires du cristallin (fig. 28-6 ).
Du mésenchyme, dérivé majoritairement des CCN mais associé à l'endothélium de capillaires d'origine mésodermique, se condense autour de la surface externe de la cupule optique [6 - 8]. La couche la plus interne de ce mésenchyme, intimement juxtaposée à la membrane basale de l'EPR, forme la membrane choroïde (lamina uvéocapillaire), un tissu conjonctif lâche et très vascularisé (fig. 28-6 et voir plus loin fig. 28-9 ). Elle est en continuité via la tige optique avec une membrane, histologiquement et fonctionnellement analogue, qui tapisse la face externe du cerveau antérieur, les méninges. En effet, la membrane choroïde est homologue dans son origine embryonnaire de la pie-mère et de l'arachnoïde, qui enveloppent le cerveau antérieur [7]. Par ailleurs, la couche externe du mésenchyme condensé autour de la cupule optique forme la sclérotique, en continuité avec la dure-mère qui enveloppe l'ensemble du cerveau antérieur. À ce titre, il est important de souligner que les cellules cartilagineuses dont la différenciation forme l'orbite squelettique de l'œil, sont également issues des CCN [2]. Sur le plan ontogénique, l'œil et le cerveau antérieur bénéficient par conséquent d'un soutien vasculaire et squelettique dérivé des CCN; plus caudalement, c'est le mésoderme qui fournit la majeure partie du mésenchyme vasculaire et squelettique annexé au système nerveux central et périphérique.
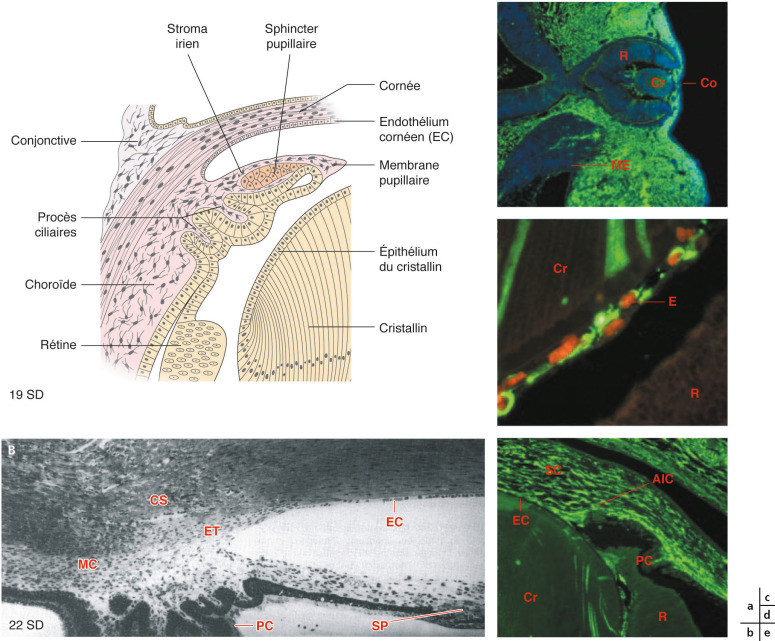
Fig. 28-6 Maturation du cristallin et de l’angle iridocornéen.
a. Illustration de 1907 de l’anatomie de l’angle iridocornéen humain, autour de 19 semaines de développement (SD). b. Photomicrographie de 1989 montrant les structures analogues chez un foetus vers 22 SD. c – e. Photomicrographies en immunofl uorescence traçant le devenir de la majorité des cellules de la crête neurale (CCN) chez des souris transgéniques. Les CCN sont en vert, les noyaux en (c) sont en bleu et les érythrocytes en (d) sont en rouge. À J10,5 sur J21 de gestation de la souris, la forme de l’oeil est similaire à l’oeil humain vers la fi n de 6 SD. Les CCN investissent l’espace entre ectoderme et cristallin pour donner la cornée, ainsi que l’espace derrière le cristallin pour participer au tissu conjonctif des vaisseaux hyaloïdes. d. Les CCN persistent en tant que péricytes de l’ensemble des vaisseaux sanguins de l’oeil à la naissance. e. L’angle iridocornéen présente un fort contingent de CCN, notamment dans le stroma cornéen, à l’exception des processus ciliaires, la rétine neurosensorielle et le cristallin. AIC : angle irido-cornéen ; Co : cornée ; Cr : cristallin (« lens crystallina ») ; CS : canal de Schlemm ; E : érythrocyte ; EC : endothélium cornéen ; ET : ébauche trabéculaire ; MC : muscle ciliaire ; ME : muscle extra-oculaire ; PC : processus ciliaires ; R : rétine ; SC : stroma cornéen ; SP : sphincter pupillae.
Syndrome de Sturge-Weber -Krabbe
Dans le syndrome de Sturge-Weber-Krabbe, l’association de déficits du réseau capillaire facial du derme périorbital (se manifestant par des « taches de vin »), d’un glaucome ipsilatéral et de calcifications épileptogènes des leptoméninges corticales est due à une mutation stéréotypée et activatrice d’un gène codant pour un relais moléculaire de signal impliqué dans la prolifération et la différenciation cellulaire [9]. Il a été montré que cette mutation apparaît de novo, après la fécondation, dans la lignée des cellules endothéliales et se révèle dans le secteur des vaisseaux dont le muscle lisse est assuré par les CCN.
Pendant la huitième et dernière semaine de la période embryonnaire, les axones des cellules ganglionnaires de la rétine progressent vers la tige optique. Ces axones s'allongent à l'intérieur de la tige puis vers le cerveau, formant ainsi le nerf optique. Parallèlement, se forment les fibres secondaires du cristallin, ainsi que les sutures de la lentille et le corps vitré secondaire.
En résumé, à l'issue de la période embryonnaire, l'œil est composé de structures épithéliales composées d'une cupule à double couche et enserrant un cristallin dérivé de l'ectoderme de surface. La cupule comporte une couche interne de neuro-ectoderme, à l'origine de la future rétine, et une couche fine externe d'épithélium pigmentaire (EPR), en continuité avec le nerf optique. L'œil comprend également un important contingent mésenchymateux périoculaire, lui-même constitué d'une couche externe dense, formant la majeure partie de la cornée et de la sclérotique, et d'une couche vasculaire lâche qui forme la choroïde, le stroma de l'iris et les corps ciliaires (fig. 28-6 et 28-7 ). À ce stade, l'embryon humain fait 3 cm de longueur et le diamètre du globe oculaire est compris entre 1,5 et 2,0 mm.
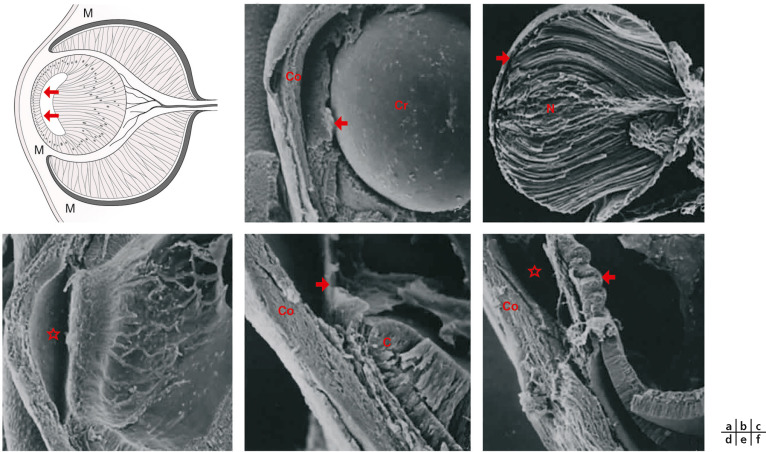
Fig. 28-7 La maturation du cristallin fait en sorte que les premières cellules, cuboïdes, ne persistent que sur la face antérieure, alors que les fibres secondaires, longitudinales, croissent pour combler la cavité. Chambre antérieure à 8 semaines de développement (SD) par microscopie électronique à balayage : le stroma de la cornée (C) est présent ainsi que la membrane pupillaire (flèche) qui recouvre le cristallin (Cr). L'intérieur du cristallin à 7 SD montre le noyau embryonnaire (N) de la structure et l'épithélium fin, antérieurement (flèche). Sous le cristallin et la membrane pupillaire, on distingue la chambre antérieure (étoile) et les vaisseaux sanguins. A plus fort grossissement, la marge antérieure de la cupule optique (C) est visible ainsi que des fentes dans la région limbique de la cornée (Co) qui se rejoignent pour former le canal de Schlemm. La flèche indique le bord de la membrane pupillaire. Vers 13 SD, les processus ciliaires (flèche) se forment dans l'iris postérieur exposant ainsi la trame trabéculaire.
(fig. 28-7 a, adapté d’un schéma de T.-C. Hengst ; fi g. 28-7b à f, remerciements au Dr K.-K. Sulik.)
Conservation fonctionnelle génétique au cours de l’évolution de l’oeil
Le chapitre 11.2 résume certains des gènes nécessaires au développement de l’oeil chez plusieurs espèces. Les lecteurs intéressés par les modèles animaux destinés à l’étude du développement de l’oeil et également par les mécanismes qui ont conduit à la diversification des fonctions de ces gènes au cours de l’évolution sont incités à consulter une édition spéciale de l’International Journal of Developmental Biology [10].
Malformations sévères du tube neural et de l’oeil
Malformations sévères du tube neural et de l’oeil
l’anophtalmie primaire, résultat d’un échec de la formation de la vésicule optique. Les orbites ne contiennent pas de tissu oculaire. Cependant, dans certaines formes d’anophtalmie, les muscles extra-oculaires issus du mésoderme, les tendons et tissus conjonctifs issus des CCN, ainsi que les glandes lacrymales issues de l’association de l’ectoderme et des CCN sont bien présents. La présence de ces dérivés atteste, par conséquent, d’un début d’induction des structures oculaires qui ont secondairement subi une dégénérescence. Cette malformation très rare est associée principalement à des mutations des gènes codant les facteurs de transcription RAX, OTX2 et SOX2, lesquels peuvent conjointement engendrer un spectre de malformations syndromiques affectant le développement du cerveau antérieur ;
la nanophthalmie et la microphtalmie correspondent, l’une et l’autre, à un défaut de croissance de l’ébauche oculaire. Dans ce cas, le développement initial est bien engagé, mais les structures oculaires cessent de croître prématurément, ce qui produit un oeil rudimentaire et hypoplasique. L’oeil qualifié de « microphtalme » est petit à la naissance mais contient des éléments reconnaissables tels qu’un cristallin, une membrane choroïde et une rétine ;
la synophthalmie correspond à la fusion des deux ébauches oculaires qui peut résulter soit d’une malformation, soit de processus inductifs défectueux, soit dépendre d’un défaut de différenciation du tissu mésenchymateux entre les vésicules optiques. Il est rare qu’un seul oeil (cyclope) se forme par ce mécanisme : dans la plupart des cas, les deux cornées et les deux cristallins sont bien individualisés, de même que les iris et les corps ciliaires correspondants. En revanche, les structures craniofaciales et les annexes oculaires médianes sont manquantes, si bien que la sclérotique de la ligne médiane et le tissu uvéal peuvent aussi être absents ; dans ce cas, le nerf optique peut être simple ou double. Cette malformation peut être associée soit à une délétion du chromosome 18, soit à une holoprosencéphalie due à des mutations du gène SHH, codant pour un facteur de croissance essentiel à l’expansion de la population de CCN céphaliques, ou de gènes codant pour les effecteurs intracellulaires de cette voie de signalisation ;
l’oeil kystique congénital se caractérise par le développement d’une structure kystique désorganisée qui empêche la morphogenèse et l’invagination du disque de la rétine.
Le disque de la rétine est destiné à se différencier en neurones rétiniens, tandis que la couche fine la plus externe de la vésicule optique est destinée à former l'EPR. D'architecture et de fonctions distinctes, ces deux couches sont néanmoins en continuité dans un angle aigu au niveau de la chambre antérieure. La transition à ce niveau s'accompagne de la différenciation de structures hautement spécialisées dans la fonction optique : c'est là que se forment l'iris, le corps ciliaire et le bord de la pupille. En raison de l'invagination de la cupule optique, la partie apicale de la rétine neurale primitive vient s'adosser à la surface apicale de l'EPR, aux dépens de l'espace intrarétinien.
À l'instar des cellules épendymaires qui couvrent les espaces ventriculaires du cerveau, les cellules qui tapissent les surfaces juxtaposées de la rétine neurale primitive et du futur EPR sont ciliées (fig. 28-8 ). La différenciation des cils de la rétine neurale revêt une importance physiologique fondamentale dans la maturation des cellules réceptrices, les cônes et les bâtonnets, et la transduction du stimulus lumineux à leur niveau. La présence de ces cils primaires sur les cellules de l'EPR est également indispensable. De nombreux gènes dont la mutation est responsable de la rétinite pigmentaire touchent à la formation des cils dans l'EPR. Parmi ceux-ci, le gène GPCR est impliqué dans la majorité des cas liés au chromosome X. La compréhension du rôle du cil primaire dans l'organisation et la signalisation épithéliale reste un sujet actuel de recherche.
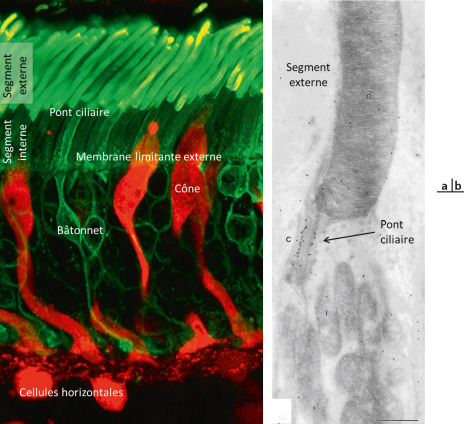
Fig. 28-8 Des mutations héritées dans des gènes codant de nombreuses protéines de la région ciliaire du segment externe sont responsables de rétinites pigmentaires isolées et syndromiques associés ou non à l’amaurose de Leber, aux syndromes d’Usher, de Bardet-Biedl et de Joubert.
(cônes et cellules horizontales, en rouge). b. Microscopie électronique à transmission pour montrer, par des points de déposition de particules d’or au pont ciliaire, la localisation de myosin 7A, dont la mutation du gène est responsable du syndrome d’Usher type B. (fig. 28-8 a, remerciements au Dr. R. Fariss, National Eye Institute ; fi g. 28-8b modifi ée de Liu X et al. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil Cytoskeleton 1997 ; 37 : 240-52.)
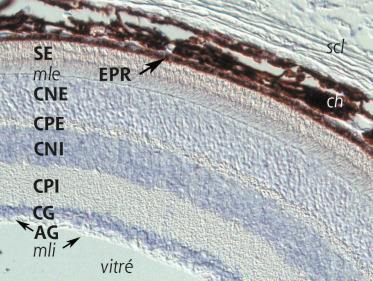
Fig. 28-9 De l’extérieur vers l’intérieur de la rétine, plusieurs couches histologiques sont identifi ables chez la souris adulte comme chez l’humain.
Couches histologiques : épithélium pigmentaire rétinien (EPR) ; couche des segments externes (SE) des photorécepteurs, en contact avec l’EPR ; segment interne au contact avec la membrane limitante externe (mle) ; couche nucléaire externe (CNE) ; couche plexiforme externe (CPE) ; couche nucléaire interne (CNI) ; couche plexiforme interne (CPI) ; couche des corps des cellules ganglionnaires (CG) ; couche des axones ganglionnaires (AG) qui convergent vers le nerf optique ; membrane limitante interne (mli).
De l'extérieur vers l'intérieur de la rétine mature, plusieurs couches histologiques deviennent identifiables (fig. 28-9 ). Elles comprennent six types de cellules nerveuses spécialisées :
- – l'épithélium pigmentaire rétinien (EPR);
- – la couche des segments externes (SE) des photorécepteurs : cônes (1) et bâtonnets (2) au contact de l'EPR;
- – la « membrane » limitante externe (MLE);
- – la couche nucléaire ou granulaire externe (CNE), contenant les corps des photorécepteurs;
- – la couche plexiforme externe (CPE), où les dendrites des cellules radiaires bipolaires (3) et les dendrites des cellules horizontales (4) intègrent les signaux des photorécepteurs;
- – la couche nucléaire ou granulaire interne (CNI), comprenant les corps des interneurones bipolaires, horizontaux et amacrines (5);
- – la couche plexiforme interne (CPI), contenant les axones de cellules bipolaires, connectés aux dendrites des cellules ganglionnaires, ainsi que les ramifications des cellules amacrines;
- – la couche des corps des cellules ganglionnaires ou CG (6);
- – la couche des axones ganglionnaires (AG) qui convergent vers le nerf optique;
- – la membrane limitante interne (MLI).
La rétine neurale primitive, à l'origine de la majorité de la rétine mature, se compose tout d'abord d'une zone nucléaire et d'une zone acellulaire intérieure. La zone nucléaire correspond au neuroépithélium ventriculaire prolifératif du tube neural, et contient des cellules multipotentes. Les couches interne et externe de la cupule optique à ce stade ont des lames basales distinctes : celle de la couche intérieure donne la MLI, et celle de la couche externe, la membrane de Bruch. La différenciation des couches neurales rétiniennes commence au pôle postérieur et progresse d'une manière centrifuge, donnant un gradient de différenciation de la rétine neurale à l'intérieur de l'œil autour de 7 sd. L'activité mitotique de la rétine neurale primitive est également plus grande dans la couche neuroblastique externe germinative. Les cellules nouvellement formées migrent vers l'intérieur de la cupule au niveau de la zone marginale pour donner la couche neuroblastique interne. Ces deux couches sont bien individualisées vers 6-7 sd par une zone cellulaire moins dense, mais également proliférative, connue sous le nom de couche transitoire de Chievitz [11]. Des études de cartographie réalisées chez de nombreux modèles vertébrés ont montré la dynamique de ce processus en suivant la fluorescence émise par une protéine transgénique, produite de façon constitutive par les clones de cellules mosaïques [12]. Cette approche a montré la structuration en colonnes et le début de la stratification de la rétine, secondairement suivis par une ramification latérale.
Sur la face interne de la couche nucléaire, les axones des cellules ganglionnaires, premiers neurones à se différencier, convergent vers la tige optique. Une zone où les processus des cellules de la couche nucléaire intérieure s'entremêlent, la couche plexiforme interne, devient identifiable, au détriment de la couche transitoire de Chievitz qui disparaît (fig. 28-9 ). Les premières cellules différenciées de la couche neuroblastique interne donnent les cellules radiaires gliales de Müller et les cellules amacrines, qui forment ainsi la couche nucléaire interne à partir du pôle postérieur de la rétine. Peu de temps après, les cellules bipolaires et horizontales se différencient dans la couche neuroblastique externe et migrent vers la nouvelle couche nucléaire interne. Les composants cellulaires restant de la couche neuroblastique extérieure forment ensuite la couche nucléaire externe, contenant les corps cellulaires des photo récepteurs (cônes et bâtonnets). La zone où les fibres de cette couche se mêlent à celles de la couche nucléaire interne constitue la nouvelle couche plexiforme externe (fig. 28-9 ). La « membrane » limitante externe, n'est pas une membrane à proprement parler, mais se manifeste par l'alignement et la densité des jonctions serrées impliquant les photorécepteurs et les cellules de Müller.
Le développement oculaire est également marqué par d'autres étapes importantes pour l'élaboration de la rétine. Elles concernent d'une part, la formation de la microglie (c'est-à-dire des macrophages tissulaires résidents), dont les cellules investissent la rétine via le système vasculaire rétinien et sous-rétinien (10–12 sd), et d'autre part, la synaptogenèse qui débute à partir des pédoncules des cônes à 4 mois de gestation, et à partir des sphérules des bâtonnets à 5 mois.
Un des événements frappants du développement de l'œil est l'apparition de la mélanine dans l'EPR embryonnaire vers J28. Initialement, l'EPR est un épithélium pseudo-stratifié cilié et mitotiquement actif. Les cils disparaissent alors que commence la mélanogenèse. Les cellules de l'EPR acquièrent une forme hexagonale et s'organisent en épithélium cubique simple, bien qu'une organisation pseudo-stratifiée se maintienne dans la rétine périphérique plus longtemps. Au cours du 4 mois, l'EPR présente des microvillosités apicales, peu ou pas de replis basaux, des interdigitations basolatérales primitives et des vésicules, les mélanosomes, qui séquestrent la mélanine dans le cytoplasme (fig. 28-10 ). L'activité mitotique a lieu très tôt dans le développement mais est, pour l'essentiel, terminée à la naissance. Par conséquent, la croissance de l'œil, et de l'EPR proprement dit, se fait par hypertrophie, c'est-à-dire par élargissement des cellules existantes. Les premières composantes de la membrane de Bruch, la lame basale de l'EPR, sont reconnaissables dès le stade de la cupule optique. Des fibrilles de collagène sont ensuite déposées sous la lame basale vers 10 sd; la première ébauche de la couche élastique peut être détectée dès 15 sd et devient fenestrée à la mi-gestation.
Malformations de la rétine
Des défauts dans l’organisation des cellules neuroblastiques de la rétine conduisent à l’épaississement et à la distorsion de l’architecture des réseaux neuronaux. Cette « dysplasie » rétinienne se manifeste par la formation de foyers de cellules neuroblastiques égarées au cours de la différenciation, formant alors des structures sphériques ou ovoïdes, appelées rosettes. Des anomalies plus subtiles peuvent survenir dans la formation des bâtonnets et des cônes qui peuvent conduire à des défauts de la vision des couleurs ou à une mauvaise acuité visuelle associée à un nystagmus congénital.
La maculogenèse se manifeste vers la mi-gestation par une augmentation localisée de la densité de cellules ganglionnaires situées au bord temporal du disque optique. À 6 mois, la couche des cellules ganglionnaires peut atteindre une profondeur de 8 à 9 cellules dans cette région. La couche nucléaire externe épaissie consiste principalement en des cônes immatures. Vers le 7 mois, un déplacement des cellules ganglionnaires vient à former une dépression fovéale. Au 8 mois, il n'y a plus que deux couches de cellules ganglionnaires à ce niveau et à la naissance, la couche est monocellulaire. Pendant les 4 mois qui suivent la naissance, la couche de cellules ganglionnaires et la couche nucléaire interne se retirent jusqu'aux marges de la fovéa, ne laissant que des cônes dans la fovéa. L'allongement des segments internes et externes se poursuit au cours des mois suivants.
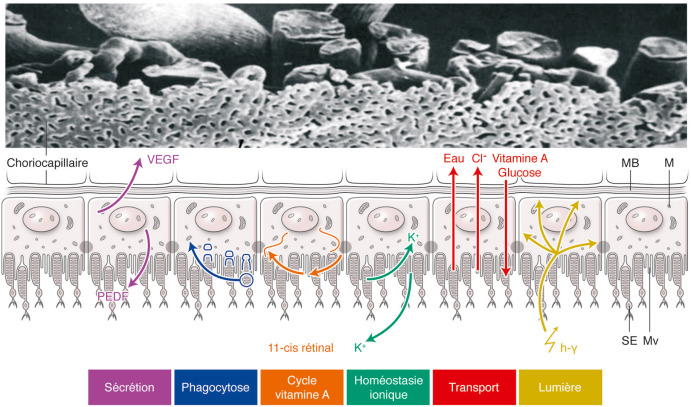
Fig. 28-10 Interactions entre la couche interne vasculaire de la choroïde, l’épithélium pigmentaire (EPR) et les photorécepteurs de la rétine.
En haut, microscopie électronique à balayage des trois couches vasculaires de la choroïde dont la plus interne, le choriocapillaris, est séparée par une lame basale du contact direct avec l’EPR. M : mélanosome ; MB : membrane de Bruch ; Mv : microvillosités : PEDF : pigment epithelium-derived factor (serpin F1) ; SE : segment externe ; VEGF : vascular endothelial growth factor. (Figure adaptée de Zhang HR. Scanning electron-microscopic study of corrosion casts on retinal and choroidal angioarchitecture in man and animals. Progress in Retinal and Eye Research 1994 ; 13 : 243-70, avec l’accord d’Elsevier et Strauss O. The retinal pigment epithelium in visual function. Physiol Rev 2005, 85 : 845-81.)
Jusqu'à 10-12 sd, la périphérie de la rétine ne s'étend pas jusqu'au bord de l'intérieur de la marge de la cupule optique. À 14 sd, elle se termine au niveau des futurs procès ciliaires, nouvellement formés. La pars plana définitive ainsi que l'ora serrata rudimentaire et la pars plicata sont présentes vers 6 mois de gestation. La pars plana et la région de l'ora serrata à l'équateur de l'œil continuent de s'étendre après la naissance avec la croissance continue du globe oculaire, qui se poursuit principalement jusqu'à 2 ans d'âge. À la naissance, la zone de la rétine est approximativement de 600 mm et atteint 800 mm vers l'âge de 2 ans.
Issue de l'artère carotide interne, l'artère ophtalmique se ramifie pour donner l'artère hyaloïde qui s'incorpore à la fissure optique (fig. 28-5 à 28-7 ). L'artère hyaloïde, après avoir émergé de l'axe de la tige optique, se ramifie entre la surface du cristallin et la zone marginale de la rétine neurale primitive (espace lentorétinal). Avec la croissance de la cupule optique et la formation de la cavité vitréenne, l'artère hyaloïde traverse le corps vitré primitif, dans le canal hyaloïde ou canal de Cloquet, afin d'atteindre la surface postérieure du cristallin. La veine hyaloïde suit le même chemin en sens inverse.
Au début du 4 mois de développement, des bourgeons angiogéniques se ramifient à partir des vaisseaux hyaloïdes sur le disque optique par division cellulaire et intercalation. Ces bourgeons sont constitués d'abord de cellules endothéliales, puis accompagnés par des péricytes qui proviennent des CCN et des macrophages (fig. 28-6 d). Les cordons initiaux de cellules endothéliales se canalisent et forment de nouveaux vaisseaux qui longent la couche de fibres nerveuses vers la rétine périphérique et progressent à une vitesse d'environ 0,1 mm par jour, pour atteindre l'ora serrata vers le 8 mois. En même temps, les bourgeons pénètrent également dans la profondeur de la rétine neurale jusqu'à la frontière extérieure de la couche nucléaire externe, selon une cinétique qui se poursuit après la naissance (fig. 28-11 ). À ce niveau, ils forment un réseau polygonal de vaisseaux, le plexus rétinien extérieur [13]. La partie de l'artère hyaloïde à l'intérieur de la rétine neurale donne l'artère centrale de la rétine. Les capillaires se rejoignent et développent des jonctions serrées mais aussi communicantes immatures, cependant leurs lames basales sont incomplètes.
Rétinopathie des prématurés
L’hypoxie et l’hyperoxie sont susceptibles d’entraîner des effets stéréotypés sur la formation de réseaux capillaires de la rétine. En effet, les protéines de réponse au stress de l’hypoxie se fixent naturellement sur les promoteurs de gènes qui favorisent l’angiogenèse. À l’inverse, si des nourrissons prématurés sont placés dans un environnement où la pression d’oxygène est élevée, comme cela se pratiquait autrefois dans les couveuses, cela peut provoquer un retard ou une réduction de la vascularisation de la rétine par régression des microvaisseaux et l’inhibition de la formation de bourgeons vasculaires. Au retour à un taux et une pression d’oxygène normaux, les tissus subissent à nouveau une hypoxie localisée dans la rétine, ce qui entraîne des épisodes de néovascularisation anormale au sein de la rétine et du vitré, connus cliniquement comme la rétinopathie des prématurés. Les grands prématurés nés à moins de 30 sd sont particulièrement à risque et peuvent dans une certaine mesure être traités par photocoagulation au laser et, plus récemment, exceptionnellement, par des anti-VEGF (vascular endothelial growth factors ou facteurs de croissance de cellules endothéliales vasculaires). La rétinopathie des prématurés reste un grand problème de santé publique dans le monde [14, 15].
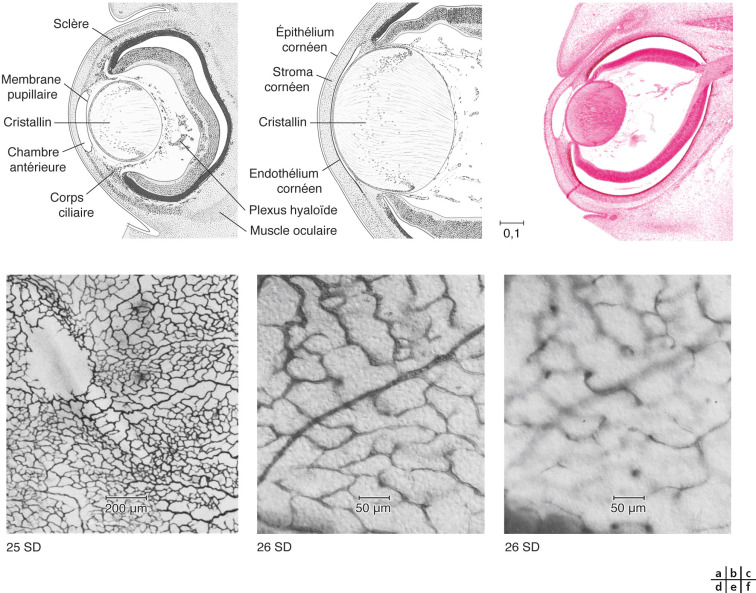
Fig. 28-11 La vascularisation de la rétine en profondeur commence par le plexus rétinien intérieur qui se développe à partir des vaisseaux hyaloïdes à la fi n de 8 semaines de développement (SD).
à l’éosine. d. Vers 25 SD, au niveau de la fovéa. e, f. Immunohistochimie contre CD34, ce qui met en évidence les cellules endothéliales du plexus rétinien intérieur et extérieur en formation, respectivement, à 26 SD. Une artériole (a) est également visible. (fig. 28-11 a et b, reproduit de O’Rahilly R, Müller F. [27], avec autorisation. fig. 28-11 c, Récupéré le 13 janvier 2017, de l’image interactive à https: //embryology.med.unsw.edu.au/embryology/Slides/Embryo_Stages/Stage22/08-eye/Stage22-08-eye.html, avec autorisation de l’auteur ; fi g. 28-11d à f, reproduit de Hughes S et al. [13], avec autorisation.)
La tige optique assure, en son centre, la communication et la circulation des fluides entre la cavité du cerveau antérieur (futur troisième ventricule du cerveau) et la cavité des vésicules optiques en cours de développement (fig. 28-4 b). Vers la fin de la 4 semaine de gestation, la tige est remplie de fluide et bordée par des cellules neuro-ectodermiques. L'invagination de la tige optique pour la formation de la fissure choroïdienne à sa face ventrale est concomitante de l'invagination de la vésicule optique en cupule optique. Pour la tige optique, ces mouvements morphogénétiques se traduisent par la formation d'une double couche de neuro-ectoderme qui se replie et s'affaisse sur elle-même : ce collapsus entraîne la disparition de la cavité intermédiaire de fluide et conduit à l'incorporation des vaisseaux hyaloïdes et du mésenchyme provenant des CCN qui les accompagne (fig. 28-4 ). Les bords de la tige optique se ferment d'abord sur les vaisseaux hyaloïdes près du cerveau vers 5-6 sd, puis leur fusion s'étend distalement pour atteindre le bord inférieur de la cupule. Par la croissance asymétrique de la rétine temporale, le nerf est déporté du côté nasal vers la fin du 1 trimestre. Chez le fœtus, les tiges optiques se situent à environ 65° par rapport au plan mi-sagittal, alors que par croissance différentielle, le nerf optique qui en est issu se situe plutôt à 40° chez l'adulte.
La tige optique conduit des axones des neurones ganglionnaires de la rétine vers le cerveau. Le neuro-ectoderme extérieur de la tige se différencie en cellules gliales qui entourent le nerf optique mais donnent aussi la composante gliale de la lame criblée [16]. À l'intérieur du nerf optique, les faisceaux axonaux sont entourés par d'autres cellules gliales myélinisantes qui se sont différenciées à partir de la couche interne de la tige optique [17]. Comme pour le reste du diencéphale, le mésenchyme dérivé des CCN adjacent fournit les composantes conjonctives et méningées du nerf optique.
Malformations de la tête du nerf optique
Lorsqu’un défaut de la fermeture de la fissure optique survient dans sa partie postérieure, il peut être à l’origine d’un colobome de la tête du nerf optique. Les colobomes touchant le nerf optique peuvent être isolés ou associés à des colobomes choriorétiniens. Situées dans la partie inféronasale, ces malformations peuvent être associées à un staphylome, c’està- dire une hernie de la sclérotique. De même, elles peuvent impliquer une hernie rétinienne dans les méninges du nerf optique, parfois sous la forme d’une simple dépression ou fossette localisée au bord du disque. L’appellation historique de morning glory syndrome, qu’on retrouve parfois dans la littérature, est due à l’aspect qu’évoquent son observation au fond d’oeil et sa ressemblance avec la fleur de liseron ou ipomée. L’importance clinique de ces manifestations tient aux complications qu’elles peuvent provoquer, notamment l’infiltration de fluide sous la macula, susceptible d’entraîner le décollement de cette dernière. D’autres formes présentent une excavation très importante de la papille et une leucocorie évidente. Du tissu adipeux, du muscle lisse ou des cellules gliales ectopiques peuvent être présents à l’intérieur des méninges, ainsi que des hernies correspondantes du cerveau [18]. Les formes sévères et syndromiques peuvent s’accompagner d’agénésie du corps calleux ou de malformations de l’axe hypothalamo-hypophysaire [19].
L'ectoderme de surface, qui recouvre son intégrité après individualisation de la vésicule cristallinienne, est destiné à former l'épithélium cornéen, stratifié par quelques couches cellulaires. Une fois l'épithélium cornéen formé, vers 5 sd, les CCN du mésenchyme périoculaire migrent le long du bord de la cupule optique pour investir l'espace, compris entre l'ectoderme et la surface antérieure du cristallin, et constituer une couche oligocellulaire, l'endothélium cornéen. Cet endothélium lui-même repose sur une lame de matrice basale qui préfigure la membrane de Descemet. Vers 7 sd, une deuxième vague de CCN vient s'infiltrer entre l'endothélium et l'épithélium cornéen pour former le stroma cornéen, au départ un mésenchyme lâche, mais qui, à mesure que ces cellules se différencient, tend à se stratifier (fig. 28-6 et fig. 28-11 ).
Dans les semaines qui suivent, les paupières se forment à partir d'une expansion de l'ectoderme périoculaire, soutenue par l'accumulation du mésenchyme sous-jacent provenant des CCN. Ensuite, les paupières se soudent vers 9-10 sd et restent fermées pendant plusieurs mois (fig. 28-11 ). Vers la mi-gestation, toutes les couches de la cornée sont présentes, à l'exception de la « membrane » de Bowman, qui est une couche épaisse de collagène acellulaire sous l'épithélium cornéen. Les faisceaux de collagène dans le stroma s'organisent en lamelles fasciculées, et les cellules stromales fibroblastiques, alors désignées comme des kératocytes, s'aplatissent et développent une intense activité sécrétrice de protéoglycanes notamment. La cornée reçoit un important contingent d'axones sensoriels provenant du ganglion trigéminal (fig. 28-12 a et b) qui intéresse d'abord la périphérie de la cornée, mais qui s'étend de façon centripète entre le 3 et le 5 mois [20]. Le processus de maturation de la cornée est initié dans les couches les plus profondes de la cornée, puis progresse plus superficiellement. Une vague de différentiation des CCN, qui vont constituer la majeure partie de la cornée, les nerfs sensoriels et la sclère, précède cette maturation [21]. La composition des glycosaminoglycanes, tel l'acide hyaluronique, change au cours du temps. Cette fluctuation permet de favoriser l'hydratation et le gonflement de la gelée matricielle pendant la période où les paupières sont fusionnées, puis sa compaction à l'ouverture de ces dernières, à partir de la 24 semaine [22]. Ces changements s'accompagnent d'un arrêt quasi complet de la prolifération cellulaire au sein de la cornée, la vie durant.
Malformations congénitales de la cornée
Lorsque la membrane basale qui sous-tend l’endothélium cornéen, la membrane de Descemet, présente à sa périphérie – désignée par la ligne ou l’anneau de Schwalbe – un épaississement anormal au point qu’il devienne proéminent et ectopique, on parle d’embryotoxon (du grec « toxon », un arc). Cette malformation est visible par l’examen de l’angle iridocornéen en gonioscopie et affecterait près d’une personne sur six dans la population. L’anomalie d’Axenfeld regroupe l’embryotoxon postérieur avec des adhésions ou synéchies iridocornéennes périphériques. Si celles-ci sont associées à un glaucome et/ou d’autres malformations variables telles que la microcornée, l’hypoplasie de l’iris, la polycorie (pupilles multiples), la correctopie (pupille excentrée), des dysmorphies faciales (hypoplasie maxillaire, malformations dentaires), il s’agit alors du syndrome d’Axenfeld-Rieger. La combinaison d’adhésions iriennes à la face postérieure de la cornée et de l’opacification cornéenne correspond à l’anomalie de Peters, souvent étroitement associée au développement d’un glaucome (voir chapitres 11 et 12). Ces malformations sont génétiquement hétérogènes : elles font partie d’un spectre de mutations qui affectent l’activité des gènes codant pour des facteurs de transcription nécessaires au développement précoce de l’oeil chez tous les Vertébrés.
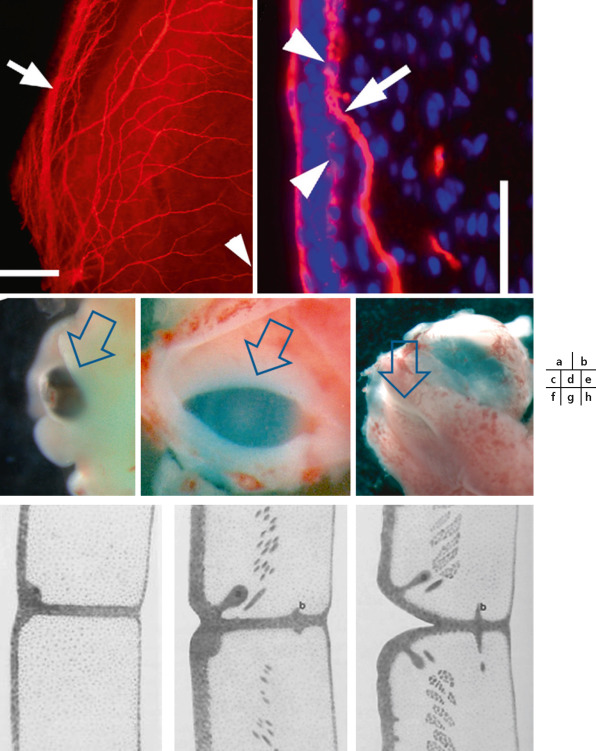
Fig. 28-12 Maturation de la cornée et des paupières.
a. La cornée reçoit un important contingent de fi bres sensorielles provenant du ganglion trigéminal, tout d’abord aux bords périphériques de la cornée, mais qui s’étend de façon centripète entre le 3e et 5e mois. b. Le processus de maturation de la cornée est initié dans les couches les plus profondes de la cornée, puis progresse plus superfi ciellement. c. À partir de 6 semaines de développement (SD), les paupières se forment à partir d’une expansion de l’ectoderme. d. Elles se ferment vers 9–10 SD. e. Les paupières restent soudées pendant plusieurs mois. f. Début de formation des follicules ciliaires. g. Le développement des follicules des cils se poursuit, alors que des invaginations postérieures signalent le début de la formation des glandes de Meibomius et le muscle orbicularis se différencie au centre. h. La progression de la différenciation ciliaire et musculaire se poursuit. La disjonction des paupières débute antérieurement vers 20 SD. (fig. 28-12 a, b, adapté de [20] ; fi g. 28-12f, h : adapté de [26], avec l’accord de Wiley.)
La placode cristallinienne s'épaissit dans l'ectoderme en regard des vésicules optiques vers J27. La différenciation du cristallin requiert la mise en jeu de deux types de signaux protéiques échangés entre les contingents cellulaires présents, ainsi que la compétence de la placode à y répondre : d'une part, un signal inductif, produit par le neuro-épithélium, et d'autre part, un signal répressif du mésenchyme provenant des CCN, qui permet de circonscrire la formation de cristallin à un endroit précis, en inhibant et contrecarrant le potentiel cristallinien de l'ectoderme environnant.
Après l'induction de la placode, l'ectoderme cristallinien s'invagine en se renfermant sur lui-même, afin de former une vésicule creuse. L'individualisation du cristallin de l'ectoderme de surface vers J33 marque le moment où la chambre antérieure commence à se façonner. À la face postérieure de la vésicule cristallinienne, les cellules organisées en couche simple tendent à se différencier sous l'effet inducteur de la rétine [23] et débutent une élongation qui les conduit à croître vers la lumière de la vésicule cristallinienne et en direction de l'ectoderme (fig. 28-7 ); cette étape d'élongation est indispensable à l'acquisition du pouvoir réfractif du cristallin [5].
Malformations du cristallin
La juxtaposition de la vésicule optique et de l’ectoderme compétent est absolument requise pour permettre l’induction d’un cristallin et son bon positionnement. Les perturbations précoces de ce processus conduisent à l’aphaquie primaire congénitale (du grec phakos, lenti lle), c’est-à-dire l’absence de cristallin. De même, la taille de la placode cristallinienne dépend du contact initial entre la vésicule optique et l’ectoderme. Ainsi, une petite placode amènera la microphaquie. Toutefois, ces malformations surviennent rarement de façon isolée et sont souvent associées à des anomalies de la chambre antérieure telles que des dysgénésies du segment antérieur. Plusieurs malformations sont le résultat d’une séparation défectueuse de ces tissus à des stades plus tardifs. À titre d’exemple, il s’agit :
des adhésions kératolenticulaires typiques de l’anomalie de Peters ;
du lenticône, qui se manifeste par une protrusion antérieure du cristallin provoquée par un manque de consistance de sa membrane basale, dû à l’absence d’un de ses composants en collagène, notamment dans le syndrome d’Alport ;
de la cataracte sous-capsulaire antérieure provoquée par une opacification de cette membrane basale qui s’épaissit dans la partie antérieure, au cours du temps.
Des anomalies rares telles qu’une duplication de la placode optique ont été rapportées. Cela entraîne la formation de deux cristallins (fig. 28-13 ).
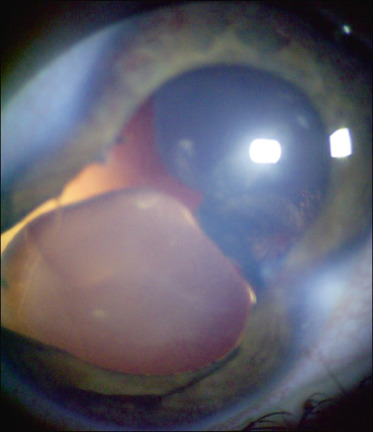
Fig. 28-13 Duplication de la placode optique entraînant la présence de deux cristallins.
(Remerciements Pr Solange Milazzo.)
Le vasa hyaloidea et la tunique vasculaire du cristallin (ou tunica vasculosa lentis) désignent un abondant réseau capillaire issu de l'artère hyaloïde qui pénètre l'espace du vitré primitif à travers la fissure choroïde et la tige optique, pour gagner la face postérieure et latérale du cristallin. À leur niveau, les vaisseaux sont dotés d'un endothélium non fenestré accompagné d'une couche périvasculaire simple adossée à une membrane basale. Ce réseau capillaire hyaloïde destiné à assurer la vascularisation du cristallin n'a qu'une existence temporaire, le temps que se façonnent les corps ciliaires et le canal de Schlemm dans la chambre antérieure. Le vasa hyaloidea et la tunique vasculaire du cristallin disparaissent par un processus physiologique de thrombose qui laisse alors le corps vitré secondaire ou définitif, hyalin, avasculaire et acellulaire.
Malformations du vitré
La persistance anormale de la tunique vasculaire du cristallin est responsable d’une malformation congénitale considérée comme une persistance du vitré primitif [24]. Cette anomalie, généralement unilatérale, entraîne l’opacification du cristallin, et peut être associée à une élévation de la pression intra-oculaire, ainsi qu’à une microphtalmie. La masse de tissu fibreux/glial au niveau de la tête du nerf optique est connue sous le nom de papille de Bergmeister et représente le vestige glial des vaisseaux hyaloïdes incomplètement atrophiés.
L'uvée désigne un complexe vasculopigmentaire situé en position intermédiaire dans l'œil. Elle forme un continuum structural et fonctionnel qui s'étend de la partie antérieure à la région postérieure, en comprenant l'iris et les corps ciliaires (uvée antérieure) et la membrane choroïde (uvée postérieure). Cette structure assure par son réseau capillaire une fonction de soutien métabolique et de nutrition pour l'iris et les corps ciliaires. Elle participe également à la fonction visuelle par l'absorption et la limitation de la réflexion lumineuse sur la rétine, ce qui favorise un bon contraste visuel.
Comme nous l'avons déjà évoqué, la membrane choroïde est une toile vasculaire qui épouse la face externe de la rétine pigmentaire (fig. 28-10 ). Elle est juxtaposée à cette dernière par la membrane de Bruch qui forme à ce niveau une lame basale riche en collagène et en fibres élastiques. La choroïde est classiquement décrite comme une succession de couches concentriques, dont la membrane de Bruch constitue la strate la plus interne. Extérieurement, elle est limitée par la lamina suprachoroidea, composée d'un réseau de fibres élastiques avasculaires, mais riches en mélanocytes. Encadré par ces deux membranes, le réseau vasculaire du stroma de la choroïde présente une organisation topographique coaxiale selon le diamètre de ses vaisseaux, allant de la choriocapillaris – les capillaires choroïdes – en face interne, à la couche de Slatter, en position intermédiaire et composée de vaisseaux de taille moyenne, puis la couche de Haller, plus périphérique et formée de vaisseaux de diamètre plus large. La choroïde est absolument requise pour l'équilibre homéostasique des structures auxquelles elle est adossée : la rétine pigmentaire et la sclérotique, postérieurement, et l'iris et les corps ciliaires, antérieurement. Elle reçoit un soutien essentiel des CCN qui forment les péricytes qui doublent l'endothélium de ces vaisseaux ainsi que les cellules pigmentaires qui les accompagnent.
Le développement du corps ciliaire présente des similitudes avec le développement de l'iris (fig. 28-6 ). Il s'agit de la juxtaposition de l'épithélium pigmenté, auquel sont appendues les fibres zonulaires qui sous-tendent le cristallin, et du tissu musculoconjonctif dérivé des CCN, qui sont impliquées dans la production et la sécrétion de l'humeur aqueuse. L'épithélium ciliaire est marqué par 70 à 75 replis qui assurent l'insertion des fibres zonulaires. La production aqueuse débute dès 20 semaines et coïncide avec des changements concomitants dans l'angle iridocornéen.
Le muscle ciliaire, formé des fibres musculaires lisses orientées longitudinalement, se termine dans la région trabéculaire. Les fibres du muscle ciliaire circulaires ou radiales se différencient beaucoup plus tard au cours du développement, puisque leur formation n'est pas toujours totalement achevée jusqu'à environ 1 an d'âge.
L'iris constitue le diaphragme de la pupille. Il comporte les muscles lisses de l'iris, le sphincter, qui rétrécit la pupille, et les deux muscles dilatateurs de la pupille. Il s'agit de structures remarquables du fait de leur origine embryologique, seuls exemples dans l'organisme où les structures musculaires striées dérivent des CCN et non pas du mésoderme. Les muscles iriens, d'action antagoniste, reçoivent une innervation parasympathique cholinergique pour le sphincter, et sympathique adrénergique pour les muscles dilatateurs, dont la maturation s'opère en fin de gestation.
Un abondant tissu conjonctif est associé aux muscles iriens. Dérivé comme ces derniers des CCN, il renferme les mélanocytes responsables de la couleur de l'iris. La pigmentation de l'iris est initiée au cours du 4 mois de gestation et s'opère de la périphérie vers le centre, jusqu'à la naissance et dans les mois qui suivent. Il peut cependant encore évoluer pendant les premières années en fonction de l'épaisseur du stroma. Les yeux foncés laissent passer la coloration de l'épithélium du fond de l'iris à la naissance, pour céder la place aux mélanocytes étoilés pigmentés au sein du stroma. En revanche, la couleur des yeux clairs résulte de l'interférence et de la réflexion de la lumière directement sur les fibres de collagène du stroma moins pigmenté.
Aniridie
L’aniridie, rarement isolée, est généralement associée à une malformation de la chambre antérieure et plus particulièrement de l’angle iridocornéen. Cette malformation correspond à un iris histologiquement anormal et hypoplasique avec un stroma hypercellulaire, souvent associé à une prolifération aberrante de l’épithélium pigmentaire, combinée avec une anomalie ou une hypoplasie du système d’écoulement de l’humeur aqueuse. Cette anomalie coïncide également avec des opacités ou des ectopies du cristallin et une hypoplasie du nerf optique. Compte tenu de l’importance développementale et physiologique du rôle qu’exerce la crête neurale à ce niveau, cette malformation est considérée comme une « neurocristopathie » c’est-à-dire une pathologie propre aux CCN. Cette malformation génétique est le résultat de mutations du gène PAX6 (ou de son absence en cas de délétion chromosomique) qui code pour un facteur de transcription particulièrement important au fil de l’évolution de l’oeil.
Une autre particularité des compétences de la crête neurale au niveau de l'angle iridocornéen concerne la formation du canal de Schlemm, qui assure le drainage de l'humeur aqueuse et est, par conséquent, essentiel à l'homéostasie de la chambre antérieure. Cette structure est composée d'un épithélium simple et contractile, exclusivement formé des CCN. Elle constitue l'extrémité « aveugle » d'un vaisseau de type lymphatique, qui par l'activité pulsatile de son endothélium draine l'humeur aqueuse vers les veines aqueuses et épisclérales. De cette manière, le canal de Schlemm maintient une pression constante dans la chambre antérieure. Les altérations de la capacité de résorption du liquide par ce canal entraînent un glaucome.
Glaucome congénital
Un glaucome congénital peut être généré par la présence obstructive ou la persistance anormale de la « membrane » de Barkan. Il ne s’agit pas d’une membrane, proprement dite, mais d’un artefact, d’un tissu compacté. Une couche apparemment continue est en fait un artefact histologique des cellules endothéliales qui se forme à la surface du réseau trabéculaire [25]. Au cours du développement, pendant le 4e mois de gestation, ces cellules ne couvrent qu’un tiers voire la moitié de la région trabéculaire et sont déjà discontinues pour permettre une communication entre la chambre antérieure foetale et les espaces intertrabéculaires. Cependant, dans la situation pathologique où cette membrane, ce tissu compacté, persiste au niveau de l’angle iridocornéen, il existe un obstacle à la résorption de l’humeur aqueuse qui entraîne une hypertonie génératrice d’une distension du globe oculaire et de ses conséquences sur la cornée, le segment antérieur et le nerf optique (voir chapitre 12).
Il est important de garder à l'esprit qu'à la face antérieure de l'œil, l'ectoderme, situé à la périphérie du territoire dévolu à la formation de l'épithélium cornéen, est impliqué dans la formation d'annexes périoculaires non moins essentielles à la protection et à l'homéostasie de l'œil. Il s'agit du complexe palpébral qui comprend des enveloppes protectrices motiles, les paupières, ainsi que les glandes qui sécrètent le film lacrymal à la surface du globe oculaire. Cet ensemble est façonné, au cours des 2 et 3 mois de gestation, par des interactions multiples entre l'ectoderme de surface et le mésenchyme sous-jacent issu des CCN.
Les paupières naissent d'un repli de l'ectoderme et apparaissent telles des excroissances paires qui encadrent la partie supérieure et inférieure de la cornée (fig. 28-12 c à e). Les CCN forment à ce niveau la musculature striée des muscles orbiculaires, droits et releveurs de la paupière, ainsi que les muscles de Müller et la composante lisse du plan fibro-élastique. Les myofibres issues de la différenciation des CCN sont dotées d'une innervation réflexe, mais aussi volontaire. Quant au complexe glandulaire des paupières (fig. 28-12 f à h), il se forme par de multiples invaginations de l'ectoderme qui génère un ensemble de glandes lacrymales vers la fin du 4 mois de gestation [26]. Il s'agit notamment des glandes lacrymales principales qui s'ouvrent à la surface du tissu conjonctival et du cul-de-sac et sont responsables de larmes réflexes qui concourent à maintenir l'asepsie et l'hydratation de la surface oculaire. Quant aux larmes basales, elles sont produites par les glandes lacrymales accessoires, qui contribuent chacune de façon variée à la composition et au renouvellement du film lacrymal. Les glandes lacrymales accessoires tapissent le tissu conjonctival et sont responsables des sécrétions muciniques qui composent la couche profonde du film lacrymal, directement à la surface de la cornée. Les glandes sébacées de Meibomius se développent le long du tarse palpébral, à la face interne de la paupière et s'y ouvrent pour déposer une sécrétion aqueuse intermédiaire. Les glandes sudoripares de Moll sont situées à l'apex de la paupière et à proximité du cil, dont la sécrétion lipidique forme la couche la plus superficielle du film lacrymal.
La classification des malformations congénitales ou anomalies du développement est difficile pour plusieurs raisons. Tout d'abord, l'étiologie est souvent inconnue, même quand une cause génétique ou environnementale unique est suspectée. Il est par conséquent souvent difficile d'attribuer une responsabilité exclusive de ces agents ou des événements dans ces processus pathologiques, notamment parce que les causes sont souvent multifactorielles et mettent en jeu des facteurs de prédisposition. De plus, l'exposition à des agents tératogènes, tels que des médicaments ou des traumatismes, peut également entraîner des défauts de développement similaires à ceux provoqués par des accidents génétiques, en interagissant avec l'activité des gènes du développement. On peut citer, par exemple, les anomalies chromosomiques, touchant des gènes codant les nombreux facteurs de transcription qui régissent l'oculogenèse.
Ces questions mettent en relief tout l'intérêt des études en biologie du développement qui reposent sur la mise au point et l'exploitation de modèles expérimentaux, où l'analyse des relations épistatiques entre les gènes permet, d'une part, l'élaboration des réseaux génétiques impliqués, l'étude de leur cinétique d'action, et d'autre part, l'identification de facteurs de convergence comme cible thérapeutique potentielle.
[1] Slack JMW Egg & Ego : an almost true story of life in the biology lab: Springer Science & Business Media ( 2012 )
[2] Le Douarin N, Kalcheim C The ceural nrest: Cambridge University Press ( 1999 )
[3] Chao JR, Bronner ME, Lwigale PY Human fetal keratocytes have multipotent characteristics in the developing avian embryo Stem Cells Dev ( 2013 ) : 22: 2186-2195
[4] Creuzet S, Vincent C, Couly G Neural crest derivatives in ocular and periocular structures Int J Dev Biol ( 2005 ) : 49: 161-171
[5] Cvekl A, Ashery-Padan R The cellular and molecular mechanisms of vertebrate lens development Development ( 2014 ) : 141: 4432-4447
[6] Le Lievre CS, Le Douarin NM Mesenchymal derivatives of the neural crest : analysis of chimaeric quail and chick embryos J Embryol Exp Morphol ( 1975 ) : 34: 125-154
[7] Etchevers HC, Vincent C, Le Douarin NM, Couly GF The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain Development ( 2001 ) : 128: 1059-1068
[8] Gage PJ, Rhoades W, Prucka SK, Hjalt T Fate maps of neural crest and mesoderm in the mammalian eye Invest Ophthalmol Vis Sci ( 2005 ) : 46: 4200-4208
[9] Shirley MD, Tang H, Gallione CJ Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ N Engl J Med ( 2013 ) : 368: 1971-1979
[10] Bailey TJ, El-Hodiri H, Zhang L Regulation of vertebrate eye development by Rx genes Int J Dev Biol ( 2004 ) : 48: 761-770
[11] Smirnov EB, Puchkov VF Characteristics of cellular proliferation in the developing human retina Neurosci Behav Physiol ( 2004 ) : 34: 643-648
[12] Reese BE, Necessary BD, Tam PPL Clonal expansion and cell dispersion in the developing mouse retina Eur J Neurosci ( 1999 ) : 11: 2965-2978
[13] Hughes S, Yang H, Chan-Ling T Vascularization of the human fetal retina : roles of vasculogenesis and angiogenesis. Investig Ophthalmol Vis Sci ( 2000 ) : 41: 1217-1228
[14] Fierson WM, American Academy of Pediatrics Section on Ophthalmology , American Academy of Ophthalmology , American Association for Pediatric Ophthalmology and Strabismus , American Association of Certified Orthoptists Screening examination of premature infants for retinopathy of prematurity Pediatrics ( 2013 ) : 131: 189-195
[15] Gilbert C, Fielder A, Gordillo L, International NO-ROP Group Characteristics of infants with severe retinopathy of prematurity in countries with low, moderate, and high levels of development : implications for screening programs Pediatrics ( 2005 ) : 115: 518-525
[16] Hayreh SS Structure of the Optic Nerve Ischemic optic neuropathies: Springer ( 2011 ) 7-34
[17] Ye H, Hernandez MR Heterogeneity of astrocytes in human optic nerve head J Comp Neurol ( 1995 ) : 362: 441-452
[18] Mullaney J Complex sporadic colobomata Br J Ophthalmol ( 1978 ) : 62: 384-388
[19] Risse JF, Guillaume JB, Boissonnot M, Bonneau D Un syndrome polymalformatif inhabituel : l'« association CHARGE » à un « morning glory syndrome » unilatéral Ophtalmologie ( 1989 ) : 3: 196-198
[20] Marfurt CF, Cox J, Deek S, Dvorscak L Anatomy of the human corneal innervation Exp Eye Res ( 2010 ) : 90: 478-492
[21] Lwigale PY, Conrad GW, Bronner-Fraser M Graded potential of neural crest to form cornea, sensory neurons and cartilage along the rostrocaudal axis Development ( 2004 ) : 131: 1979-1991
[22] Zieske JD Corneal development associated with eyelid opening Int J Dev Biol ( 2004 ) : 48: 903-911
[23] McAvoy JW, Chamberlain CG, de Iongh RU Lens development Eye ( 1999 ) : 13: 425-437
[24] Roche O, Keita Sylla F, Beby F Persistance et hyperplasie du vitré primitif J Fr Ophtalmol ( 2007 ) : 30: 647-657
[25] McMenamin PG Human fetal iridocorneal angle : a light and scanning electron microscopic study Br J Ophthalmol ( 1989 ) : 73: 871-879
[26] Hamming N Anatomy and embryology of the eyelids : a review with special reference to the development of divided nevi Pediatr Dermatol ( 1983 ) : 1: 51-58
[27] O'Rahilly R, Müller F : Carnegie Institution of Washington ( 1987 )
C. Speeg-Schatz
La fonction visuelle, qui ne se limite pas à la simple acuité visuelle, se développe autour de deux axes :
- – l'un sensoriel centré sur l'acuité visuelle et la binocularité;
- – l'autre moteur qui est la fonction volontaire de regarder dépendant du système oculomoteur, de la maturation fovéolaire, de l'attention visuelle et du système neurologique.
Ainsi, la fonction visuelle s'articule autour de l'œil capteur ou effecteur, mais c'est le traitement cognitif de l'information sensorielle qui permet d'adapter son comportement et son regard. C'est pourquoi les fonctions visuelles sont en interaction avec notre environnement, et notre regard doit assurer en permanence l'adaptation de la réalisation d'un geste en fonction des données spatiotemporelles.
Chez le mammifère supérieur, le développement anatomofonctionnel des structures oculaires, des voies visuelles et des zones cérébrales impliquées dans la fonction visuelle sensorielle et motrice n'est pas achevé à la naissance. La maturation visuelle se déroule essentiellement durant la première année de vie [1], mais se poursuit pendant la première décennie.
Une altération de l'expérience visuelle peut entraîner une amblyopie au cours de la période sensible du développement visuel. La réversibilité des altérations des propriétés des neurones visuels, lorsque l'expérience visuelle normale est rétablie, n'est possible qu'avant une date marquant la fin de la période sensible [2].
Enfin, la fonction visuelle peut être altérée par de nombreuses pathologies pouvant résulter d'une atteinte organique de l'œil à l'origine de la malvoyance, voire de la cécité, et/ou de l'atteinte des circuits neurologiques responsables de troubles neurovisuels et compromettant de nombreux apprentissages notamment scolaires.
Le globe oculaire, qui a une longueur axiale d'environ 23 mm chez l'adulte, est entouré de trois membranes : la sclère protectrice, la choroïde vasculaire et la rétine nerveuse où est située la macula centrée par la fovéa, zone de vision fine. Nous disposons de deux types de photorécepteurs :
- – les cônes concentrés dans la fovéa et dont l'allongement et la concentration assurent la maturation de cette dernière et de ce fait la qualité de la fixation et de l'acuité visuelle; les cônes reçoivent les informations visuelles de la partie centrale du champ visuel, assurent la vision des détails, le contraste et la vision des couleurs;
- – les bâtonnets : ils permettent la vision en moyenne et faible luminance, la perception du mouvement et des formes, c'est-à-dire le champ visuel périphérique.
Les globes oculaires sont reliés au cerveau par les nerfs optiques qui se réunissent dans le chiasma optique et se prolongent par les bandelettes optiques jusqu'aux corps géniculés latéraux dorsaux qui projettent au cortex visuel primaire (aire 17 ou V) via les radiations optiques.
Ainsi, la voie optique prend son origine au niveau de la rétine de réception grâce aux photorécepteurs, se poursuit par un étage de transmission, d'abord par une voie intrarétinienne (cellules bipolaires, cellules ganglionnaires), puis par une voie extracérébrale et enfin intracérébrale.
Au plan oculomoteur enfin, les globes oculaires sont munis de deux systèmes musculaires :
- – un système intrinsèque qui assure l'accommodation, c'est-à-dire la mise au point des images sur la rétine;
- – un système extrinsèque ou oculomoteur composé de six muscles, les quatre droits et les deux obliques.
Ainsi, autour de l'œil, s'organise une boucle neurovisuelle constituée par des structures cortico-sous-corticales dont les réseaux d'interconnexion s'infiltrent dans les hémisphères cérébraux et le tronc cérébral. Ceci peut expliquer la plus grande fréquence des strabismes et des troubles neurovisuels chez l'enfant cérébro-lésé ou ancien prématuré [3].
Tous les éléments impliqués dans la vision sont formés à un stade embryonnaire précoce. En effet, on constate, lors d’une amnioscopie ou d’une illumination transabdominale, que le foetus réagit à l’alternance clair/sombre, puisqu’on note une accélération de son rythme cardiaque. Dès la 16e semaine d’aménorrhée, on observe ses yeux bouger en échographie. Il existe une interaction de communication entre la mère et le foetus, qui se continuera par une interaction entre la mère et le nourrisson.
Il se produit chez l’enfant pendant les premières années de vie trois événements majeurs en ce qui concerne le développement visuel :
- - la croissance du globe oculaire ;
- - la maturation de la fovéa par allongement des articles interne
- - la maturation corticale avec entrée en fonction des transmetteurs synaptiques de l’aire 17 ou aire V1, d’où partent des efférences vers une trentaine d’aires corticales interconnectées [5, 6]. À la naissance, le nouveau-né a une maturation visuelle inachevée en particulier du fait de :
- - la maturation fovéale : lorsque cette maturation est perturbée précocement, il en résulte un nystagmus, c’est-à-dire un mouvement pendulaire du globe et non pas une fixation stable du regard ;
- - la myélinisation du nerf optique et des relations entre le cortex strié, les régions préstriées et les régions sous-corticales. C’est durant la première année de vie que l’on va observer une grande partie de la croissance du globe, la concentration des cônes fovéolaires, le développement du champ visuel attentionnel et des mouvements oculomoteurs. La fonction visuelle participe au développement général de l’enfant qui lui-même provoque un entraînement de la fonction visuelle.
La discrimination spatiale permet, grâce à un ensemble coordonné de mouvements oculomoteurs et de perception visuelle, d'élaborer l'espace environnant : le nouveau-né recherche la lumière puis suit des yeux un visage à courte distance. Vers l'âge de 4 mois, l'accommodation s'installe et permet une vision nette à certaines distances [6].
C'est la fovéa qui joue un rôle clé dans cette discrimination spatiale. Elle représente le point 0 de l'orientation sensori-motrice du système visuel, c'est-à-dire la notion du droit devant. La rétine périphérique indiquera la localisation spatiale d'un stimulus lumineux.
L'environnement est ainsi perçu comme un ensemble d'informations organisées de façon spatiale autour de la fovéa qui est le point de fixation.
Le bébé, dès les premières semaines de vie, réagit aux visages dont il perçoit les contours. Vers l'âge de 3 mois, il reconnaît le visage de sa mère, même sur une photographie ou sur un écran. Il est sensible particulièrement aux effets d'externalité d'un objet jusqu'à l'âge de 3 mois.
À la naissance, le réflexe photomoteur est présent mais lent et de faible amplitude. Entre 2 et 4 semaines apparaît le réflexe de poursuite : l'enfant est capable de suivre des yeux une personne, un objet. Entre 4 et 12 semaines apparaît le réflexe de fusion et de coordination binoculaire. Le nouveau-né est capable de coordonner la vue et l'ouïe, puis la vue et la préhension [7].
L'acuité visuelle progresse de 1/20 à la naissance, à 4/10 à 12 mois, 10/10 vers 4 à 5 ans. À l'âge préverbal, l'acuité visuelle est impossible à apprécier de façon fiable, même si l'attirance de l'enfant par une forme structurée se détachant d'un fond uniforme avait pu être utilisée avec la méthode du regard préférentiel. Mais cette méthode n'est pas un bon test de dépistage de l'amblyopie; elle ne permet que d'évaluer une différence interoculaire comportementale [8].
La sensibilité aux contrastes est le plus faible contraste qui permet de distinguer un stimulus visuel. Le nouveau-né répond à des différences de contraste de 10 % , le nourrisson de 3 mois à des différences de contraste de 5 à 8 % et l'adulte à des différences de contraste de 2 % [9].
À l'âge de 2 mois, le champ visuel est restreint, de 30° de part et d'autre du point de fixation sur le méridien horizontal, mais son extension est très rapide, quasi achevé à la fin de la première année.
À la naissance, nous disposons de 10 % de cônes « bleus », 30 % de cônes « verts » et 60 % de cônes « rouges ». La sensibilité aux couleurs est explorable chez l'enfant par les potentiels évoqués visuels et l'électrorétinogramme chromatiques. La sensation colorée est réduite au noir et blanc avant 1 mois, puis au rouge à 1 mois, au rouge/vert à 2 mois, à toutes les couleurs vers 4 mois, sachant que la maturation totale se poursuit jusqu'à l'âge de 13 ou 14 ans.
La coopération bi-oculaire s'établit vers l'âge de 2 à 3 mois et la sommation binoculaire vers l'âge de 3 à 6 mois. En effet, chaque œil donne une image monoculaire fournie par les fovéas droite et gauche et de la fusion de ces deux images avec une petite disparité naît la perception binoculaire. La capacité de fusionner des images apparaît vers l'âge de 4 mois concomitamment à la ségrégation des colonnes de dominance corticale et de l'émergence des cellules accordées à la disparité. La fusion des images permet d'améliorer la perception tridimensionnelle par la vision stéréoscopique et achève sa maturation dans les premières années de vie. On comprendra que dans les strabismes précoces, installés dans la première année de vie, cette fonction binoculaire ne pourra jamais être obtenue [10, 11].
Les saccades réflexes ou automatiques présentes dès la naissance voient leur maturation s'achever vers l'âge de 3 ans [12].
La fixation est à la base de l'observation fine et d'échanges affectifs et sociaux précoces. Présente à la naissance, elle s'affine avec la maturation fovéolaire et l'amélioration des capacités d'attention et est acquise à la fin du premier mois.
Les saccades volontaires permettent le suivi d'une cible rapide ainsi que l'exploration; elles sont présentes à 4 mois.
La poursuite, mouvement lent initié par la fovéa, assure le maintien de la fixation sur une cible en mouvement. Au début, elle est de type saccadique, puis devient régulière à partir de l'âge de 18 mois.
Fixation et poursuite sont de bons indices du développement cortical. En effet, il existe une adaptation permanente de nos mouvements oculaires en fonction de l'environnement. Il existe de ce fait une synergie permanente entre la motricité oculaire et la motricité globale, synergie particulièrement importante durant les deux premières années de vie où s'acquiert la motricité et où le regard soutient l'équilibre, initie les mouvements et calibre les gestes.
L'acuité visuelle monoculaire ne pourra se développer qu'en fonction de la qualité de la réfraction et de la fixation, de la croissance du globe, de la position du globe oculaire, de la constitution normale de ce dernier, ainsi que des relations avec le cortex et de l'expérience visuelle. Le développement normal monoculaire de chaque œil et le développement normal de la fonction motrice permettent la binocularité normale afin de mieux « voir » et de mieux « regarder ». Tout obstacle à la formation de l'image ou à l'alignement d'un globe par rapport à un autre menace la fonction visuelle de façon irréversible si la pathologie n'est pas prise en charge pendant la période sensible du développement visuel.
[1] Hubel D. L’oeil, le cerveau et la vision : les étapes cérébrales du traitement visuel. Paris : Belin ; 1994, 240 p.
[2] Bui Quoc E. Fondements de la notion de période sensible du développement visuel. Encycl Med Chir (Elsevier, Paris). Ophtalmologie, 21-592-A-05. 2005 : 14 p.
[3] Mazeau M. Déficits visuo-spatiaux et dispraxies de l’enfant. Paris : Masson ; 1995, p. 7-32.
[4] Hendrickson AE, Yuodelis C. The morphological development of the human fovea. Ophthalmology 1984 ; 91 : 603-12.
[5] Atkinson J. Human visual development over the first 6 months of life. A review and a hypothesis. Human Neurobiol 1984 ; 3 : 61-74.
[6] Speeg-Schatz C. Développement des fonctions visuelles chez le jeune enfant. Ann Pediat 1996 ; 43 : 372-8.
[7] Atkinson J, Braddick O. Assessment of visual acuity in infancy and early childhood. Acta Ophtalmol Suppl 1983 ; 157 : 18-26.
[8] Speeg-Schatz C, Lobstein H, Flament J. Intérêt des cartons de Teller dans l’évaluation de l’acuité visuelle du jeune enfant. J Fr Ophtalmol 1991 ; 14 : 453-86
[9] Atkinson J, Braddick O, Moar K. Development of contrast sensitivity over the first 3 months of life in the human infant. Vision Res 1977 ; 17 : 1037-44.
[10] Orssaud C. Vision binoculaire. Encycl Med Chir (Elsevier, Paris). Ophtalmologie, 21-545-A-25. 2006.
[11] Atkinson J, Braddick O. Stereoscopie discrimination in infants. Perception 1976 ; 5 : 29-38.
[12] Atkinson J. Development of optokinetic nystagmus in the human infant and monkey infant : an analogue to development in kittens. In : Freemann RD. Ed. Developmental neurobiology of vision. New York : Plenum Press ; 1979.
E. Bui Quoc
Le développement des fonctions visuelles chez l'enfant est soustendu par un développement post-natal du cerveau visuel, des voies visuelles et de chaque globe oculaire. Ce développement qui débute avant la naissance au cours de la vie embryonnaire et fœtale est anatomique et fonctionnel, même si cette distinction n'est pas toujours pertinente tant les mécanismes sont intriqués. Le développement de la structure permet un développement de la fonction et dépend d'une part de facteurs acquis et de l'expérience visuelle, et d'autre part de facteurs innés avec en particulier implication de gènes du développement. Les neurones visuels acquièrent leurs propriétés de façon parallèle à la maturation de la rétine et de la fovéola en particulier; le développement sensoriel s'accompagne d'un développement moteur avec acquisition progressive de la précision des vergences, saccades et poursuites.
Les points majeurs de la croissance visuelle post-natale sont :
- – l'augmentation du diamètre cornéen;
- – l'augmentation du diamètre cornéen et la diminution de la puissance réfractive de la cornée, tous deux responsables de l'emmétropisation;
- – l'emmétropisation qui est la conséquence des deux phénomènes parallèles : augmentation de la longueur axiale et diminution de la puissance de la cornée;
- – la sagittalisation des orbites et de l'œil avec positionnement définitif des muscles autour des globes;
- – la différenciation de la fovéola;
- – la maturation des neurones du cortex visuel qui acquièrent en particulier la sensibilité à l'orientation, à la vitesse et la binocularité.
La longueur axiale de l'œil varie de 17 mm à la naissance à 23 mm à l'âge de 10 ans, avec une croissance essentiellement au profit du segment postérieur.
Fledelius et Christensen donnent des valeurs normales de 17,02 mm à la naissance, 20,19 mm à 1 an, 21,31 mm à 2 ans, 22,07 mm à 3 ans, avec un maximum de 23 mm à 10 ans [1]. Larsen retrouve des données similaires : longueur axiale de 16,78 mm à la naissance, de 18,21 mm à 6 mois, de 20,61 mm à 1 an, de 20,79 mm à 2 ans, de 21,27 mm à 3 ans, de 21,85 mm à 5 ans, de 22,50 mm à 10 ans, de 23,15 mm à 13 ans, terme de la croissance de l'œil [2-5]. Nous retenons les valeurs suivantes : 17 mm à la naissance, 18,5 mm à 6 mois, 20 mm à 1 an, 21 mm à 2 ans, 21,5 mm à 3 ans, 22 mm à 5 ans, 22,5 mm à 10 ans, 23 mm à 13 ans.
La croissance se fait au profit du segment postérieur de l'œil puisqu'on estime que la cornée à la naissance représente les trois quarts de sa surface adulte, alors que la sclère ne représente à la naissance qu'un tiers de sa surface adulte. Le volume augmente d'un facteur 2,5, puisque le volume d'une sphère [(4πR)/3] dont on prendrait comme diamètre la longueur axiale, soit 17 mm à la naissance et 23 mm à l'âge adulte, est de 2 572 mm pour un nouveau-né et de 6 370 mm à l'âge adulte. L'augmentation de volume du globe oculaire s'accompagne de l'augmentation de volume des orbites de 7 cm à la naissance à 30 cm à l'âge adulte.
Les travaux sur la croissance de la cornée après la naissance [6] nous font retenir des valeurs normales de 10,5 mm de diamètre horizontal et 10 mm de diamètre vertical à la naissance, avec augmentation jusqu'à 12 mm dans les premières années de vie : naissance = 10 mm, 6 mois = 10,5 mm, 1 an = 11 mm, 2 ans = 11,5 mm, 5 ans = 11,75 mm, > 10 ans = 12 à 12,5 mm.
La pachymétrie à plus de 580/590 µm à la naissance diminue pour atteindre 550 à 560 µm vers 10 ans, même si ce paramètre peut être constitutionnellement variable selon les individus et selon l'ethnie (pachymétrie inférieure chez le mélanoderme); la valeur dépend également de la méthode de mesure. L'étude du PEDIG rapportant ces données a porté sur plus de 2 000 enfants [7].
La puissance réfractive de la cornée est de 48 à 50 D à la naissance, 45 D entre 1 et 2 ans, 42 D à 5 ans.
La première corrélation entre structure et fonction que nous établissons est le phénomène d'emmétropisation. L'hypermétropie diminue avec l'âge avec la croissance de la longueur axiale (fig. 28-14 ). L'œil du nouveau-né n'est normalement pas hypermétrope de plus de 10 D du fait de la puissance réfractive de la cornée qui approche les 50 D [8, 9]. La forte puissance du cristallin (ou tout du moins ses capacités accommodatives faisant évaluer la puissance possible du cristallin à plus de 30 D à la naissance) contribue aussi à l'absence d'hypermétropie excessive dans la première année de vie.
Les deux phénomènes parallèles d'augmentation de la longueur axiale et de diminution de la puissance de la cornée aboutissent à l'emmétropie : sphère à 0 D et cylindre à 0 D (fig. 28-15 ).
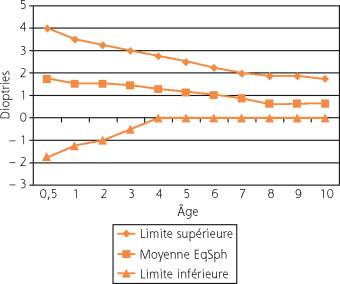
Fig. 28-14 Évolution de la réfraction chez l'enfant.
(Source : Clergeau G. La réfraction de l’enfant. ED A & J. Péchereau, Nantes, 2008. En ligne : http://www.larefraction.net/Documents/Ref-Enfant/ Ref-Enfant.html)
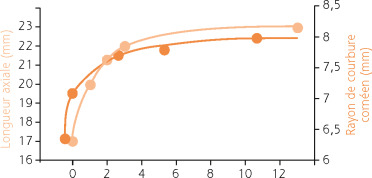
Fig. 28-15 Modifications des paramètres de la réfraction.
L’évolution de ces performances est concomitante d’une modification des paramètres biométriques du globe, que ce soit la longueur axiale, qui va passer en moyenne de 17 à 23 mm entre la naissance et l’adolescence, ou le rayon de courbure cornéen qui s’agrandit progressivement sur la même période. (D’après Hoyt C, Taylor D. Pediatric ophthalmology and strabismus. 3th Ed. Elsevier Saunders ; 2005.)
Les axes des orbites divergent de façon normale, de 35° environ chez le nourrisson et de 20° environ à l'âge adulte. Les yeux à l'état de veille sont parallèles pour une fixation de loin à l'infini, grâce au tonus musculaire. Le phénomène de sagittalisation des yeux et des orbites, au cours de l'évolution au sens darwinien du terme, et au cours du développement pré- et post-natal, est un phénomène qui permet le passage de la vision panoramique à la vision frontale stéréoscopique.
La modification de position des yeux, la croissance des globes et des orbites aboutissent à un positionnement définitif des insertions musculaires sur le globe. Classiquement, la spirale de Tillaux correspond à la localisation des insertions des muscles oculomoteurs droits, dont l'insertion par rapport au limbe recule du quadrant nasal au quadrant inférieur, puis au quadrant temporal et enfin au quadrant supérieur. Ainsi les insertions des muscles oculomoteurs se situent à une distance de 5,5 à 6 mm en nasal, de 6 à 6,5 mm en inférieur, de 6,5 à 7 mm en temporal et de 7,5 à 8 mm en supérieur. La distance du limbe des insertions des muscles oculomoteurs droits augmente avec l'âge jusqu'à 2 ans. Swan et Wilkins ont étudié cette évolution de la position des muscles oculomoteurs, avec des valeurs légèrement supérieures aux valeurs classiques, mesurant la distance à partir des extrémités des tendons et non pas du centre (plus proche du limbe du fait de la courbure du cercle cornéen) [10].
La différenciation de la fovéola dans les premiers mois et les premières années de vie est un phénomène connu depuis plusieurs dizaines d'années [11]. Cette différenciation correspond à un allongement des articles internes et externes des photorécepteurs, permettant une migration vers la périphérie des cellules bipolaires et des cellules ganglionnaires [12]. La densité des cônes au centre de la fovéola augmente de 10 000/mm à 11 semaines de grossesse, à 30 000/mm2 à la naissance et à 100 000/mm2 à 4 ans [13]. Ainsi la fovéola est mature à l'âge de 4 ans. Les études en tomographie par cohérence optique (optical coherence tomography ou OCT) permettent de démontrer anatomiquement in vivo les constatations histologiques anciennes [14].
Parallèlement à cette maturation rétinienne (maturation du capteur/récepteur), les neurones du(des) cortex visuel(s) maturent. Ils acquièrent les propriétés de sélectivité à l'orientation et à la vitesse, et des propriétés de binocularité. Ce phénomène de maturation qui peut être perturbé par une expérience visuelle anormale a été démontré dès les années 1960 par Hubel et Wiesel en électrophysiologie [15, 16] et retrouvé en imagerie optique [17, 18]. Le phénomène de développement fonctionnel des neurones visuels est retrouvé des cellules ganglionnaires aux neurones du corps géniculé latéral dorsal, jusqu'aux cellules des couches IV du cortex visuel primaire où la vision demeure monoculaire, avant de devenir binoculaire dans les couches II/III du cortex visuel primaire. Les neurones visuels maturent au niveau des cortex visuels secondaires et associatifs, de même que les neurones calleux visuels.
La période sensible du développement visuel, ou période critique, est la période de temps après la naissance pendant laquelle il existe une maturation du système visuel, et où une altération de l'expérience visuelle va entraîner des anomalies du développement et de la croissance visuelle, qui sous-tendent la question de l'amblyopie. Ces anomalies induites du système visuel peuvent être réversibles pendant la période sensible du développement visuel.
Comme les neurones sensoriels, les neurones moteurs du cortex frontal (frontal eye field ou champ oculaire frontal) et du tronc cérébral (neurones de la formation réticulée médiane [FRM] du pont ou de la formation réticulée paramédiane du pont [FRPP] ou des noyaux oculomoteurs) maturent également après la naissance, permettant d'acquérir des mouvements précis et calibrés à l'adolescence (maturation des saccades horizontales et verticales respectivement jusqu'à 6 et 12 ans; maturation des poursuites horizontales et verticales respectivement jusqu'à 10 et 15 ans).
Les fonctions visuelles du nouveau-né ne sont pas optimales et il existe donc un développement des fonctions visuelles après la naissance. C'est le développement post-natal normal des structures impliquées dans la vision, au plan sensoriel comme moteur, qui conditionne l'acquisition des fonctions normales.
Concernant l'acuité visuelle, le développement et la maturation de la fovéola jusqu'à 4 ans expliquent que la vision ne peut atteindre 10/10 ou plus qu'après cette date, de façon également dépendante de la maturation neuronale. Si à l'âge préverbal, la fonction visuelle n'est qu'estimée, soit par des tests psychophysiques fondés sur le regard préférentiel, soit par des tests objectifs comme les potentiels évoqués visuels, à l'âge verbal le caractère subjectif de l'examen doit être pris en compte dans l'évaluation de la mesure. Ceci étant souligné, on peut estimer la fonction visuelle à 1/20 à la naissance, à 1/10 vers 3 à 6 mois, à 2 à 3/10 à 1 an, à 10/10 vers 4-5 ans, et plus de 12/10 après 6 ans. Parallèlement au développement de l'acuité visuelle, l'enfant développe sa fonction de sensibilité aux contrastes, qui est présente très tôt dans les premiers mois de vie, mais qui ne mature que vers 5 à 6 ans.
Concernant la vision des couleurs, son évaluation est limitée par les contraintes d'examen, qu'ils soient subjectifs ou objectifs, de même que par la question de la culture et de l'apprentissage, la dénomination des couleurs par un enfant à l'âge verbal pouvant être difficile. Ceci dit et compte tenu de la maturation de la fovéola qui ne se termine qu'après 4 ans, et comme les cônes sont impliqués dans la vision des couleurs (opposition rouge-vert et bleu-jaune, le jaune étant un canal constitué par l'addition du rouge et du vert), on peut considérer la vision des couleurs comme possible ou normale après 4 ans.
On lit souvent que la vision binoculaire apparaît brutalement au 3 mois de vie. En fait, cette notion est sous-tendue par l'acquisition à un âge équivalent à 3 mois chez le nourrisson de la propriété de binocularité des neurones visuels du cortex visuel primaire, c'est-à-dire qu'ils peuvent être activés par la stimulation d'un œil ou de l'autre alors qu'avant cet âge ils ne sont que monoculaires. Cependant, la vision binoculaire n'est pas optimale et continue de maturer dans la première décennie de la vie, comme le montrent les études cliniques. Le test de Lang 1 vu à 2 ans et demi correspond à une stéréoscopie « grossière » : 1200″ pour le chat, 600″ pour l'étoile, 550″ pour la voiture. C'est d'ailleurs pourquoi on peut voir le test de Lang même en cas d'amblyopie. La vision binoculaire atteint 140″ à 5 ans, 80″ à 6 ans, 40″ à 9 ans, la normale de l'adulte étant 30″ [19].
Compte tenu de la position des yeux d'une part, et de la maturation et de la croissance des rétines d'autre part, le champ visuel se développe quant à lui dans les premières années de vie pour atteindre son stade quasiment mature entre 4 et 7 ans, puisqu'à ces âges on retrouve des valeurs quasiment identiques aux valeurs adultes [20] : champ visuel supérotemporal d'environ 60° à 4 ans et d'environ 70° comme chez l'adulte dès 7 ans; champ visuel inférotemporal d'environ 85° à 4, 7 et 10 ans, et de près de 95° à l'âge adulte; champ visuel inféronasal entre 50 et 55° dès 4 ans comme chez l'adulte; champ visuel supéronasal de près de 50° à 4 ans, et entre 55 et 60° dès 7 ans comme chez l'adulte.
Enfin, le système oculomoteur se met en place dans le but d'aligner les axes visuels sur la cible avec une influence du développement du système sensoriel qui a un rôle proprioceptif (copie efférente du signal), ce pourquoi le développement normal de la fonction visuelle motrice est soumis à la présence d'une expérience visuelle sensorielle normale.
La vision sensorielle et motrice d'un enfant se développe après la naissance et ce système est fragile et sensible à une expérience visuelle anormale au cours de la période critique du développement visuel qui s'achève à la fin de la première décennie en postnatal. Une anomalie de ce développement est responsable d'une amblyopie qui peut être monoculaire (moindre vision d'un œil) et/ou binoculaire (absence de vision binoculaire normale).
[1] Fledelius HC, Christensen AC Reappraisal of the human ocular growth curve in fetal life, infancy, and early childhood Br J Ophthalmol: ( 1996 ) : 80: 918-921
[2] Larsen JS The sagittal growth of the eye. 1. Ultrasonic measurement of the depth of the anterior chamber from birth to puberty Acta Ophthalmol (Copenh): ( 1971 ) : 49: 239-262
[3] Larsen JS The sagittal growth of the eye. 2. Ultrasonic measurement of the axial diameter of the lens and the anterior segment from birth to puberty Acta Ophthalmol (Copenh): ( 1971 ) : 49: 427-440
[4] Larsen JS The sagittal growth of the eye. 3. Ultrasonic measurement of the posterior segment (axial length of the vitreous) from birth to puberty Acta Ophthalmol (Copenh): ( 1971 ) : 49: 441-453
[5] Larsen JS The sagittal growth of the eye. 4. Ultrasonic measurement of the axial length of the eye from birth to puberty Acta Ophthalmol (Copenh): ( 1971 ) : 49: 873-886
[6] Ronneburger A, Basarab J, Howland HC Growth of the cornea from infancy to adolescence Ophthalmic Physiol Opt: ( 2006 ) : 26: 80-87
[7] Pediatric Eye Disease Investigator Group , Bradfield YS, Melia BM, Repka MX Central corneal thickness in children Arch Ophthalmol: ( 2011 ) : 129: 1132-1138
[8] Snir M, Friling R, Weinberger D Refraction and keratometry in 40 week old premature (corrected age) and term infants Br J Ophthalmol: ( 2004 ) : 88: 900-904
[9] Asbell PA, Chiang B, Somers ME, Morgan K Keratometry in children CLAO J: ( 1990 ) : 16: 99-102
[10] Swan KC, Wilkins JH Extraocular muscle surgery in early infancy–anatomical factors J Pediatr Ophthalmol Strabismus: ( 1984 ) : 21: 44-49
[11] Hendrickson AE, Yaodelis C The morphological development of the human fovea Ophthalmology: ( 1984 ) : 91: 603-612
[12] Sjostrand J, Popovic Z, Conradi N, Marshall J Morphometric study of the displacement of retinal ganglion cells subserving cones within the human fovea Graefes Arch Clin Exp Ophthalmol: ( 1999 ) : 237: 1014-1023
[13] Provis JM, Penfold PL, Cornish EE Anatomy and development of the macula : specialisation and the vulnerability to macular degeneration Clin Exp Optom: ( 2005 ) : 88: 269-281
[14] Vajzovic L, Hendrickson AE, O'Connell RV Maturation of the human fovea : correlation of spectral-domain optical coherence tomography findings with histology Am J Ophthalmol: ( 2012 ) : 154: 779-789 e2
[15] Hubel DH, Wiesel TN Receptive fields and functional architecture of monkey striate cortex J Physiol: ( 1968 ) : 195: 215-243
[16] Hubel DH, Wiesel TN Effects of varying stimulus size and color on single lateral geniculate cells in Rhesus monkeys Proc Natl Acad Sci U S A: ( 1966 ) : 55: 1345-1346
[17] Ribot J, Aushana Y, Bui Quoc E, Milleret C Organization and origin of spatial frequency maps in cat visual cortex J Neurosci: ( 2013 ) : 33: 13326-13343
[18] Smith GB, Sederberg A, Elyada YM The development of cortical circuits for motion discrimination Nat Neurosci: ( 2015 ) : 18: 252-261
[19] Romano PE, Romano JA, Puklin JE Stereoacuity development in children with normal binocular single vision Am J Ophthalmol: ( 1975 ) : 79: 966-971
[20] Wilson M, Quinn G, Dobson V, Breton M Normative values for visual fields in 4 – to 12-year-old children using kinetic perimetry J Pediatr Ophthalmol Strabismus: ( 1991 ) : 28: 151-153 discussion 154
I . Meunier, C. Blanchet, P. Blanchet, A. Lacroux, C. -M. Dhaenens, A. -F. Roux, B. Bocquet, C. Hamel
Les maladies oculaires héréditaires restent rares et seront évoquées après avoir éliminé une cause infectieuse, inflammatoire, tumorale, traumatique, toxique ou paranéoplasique (rarissime chez l'enfant). Nous aborderons ici les particularités et les difficultés du conseil génétique appliqué à notre pratique de la génétique des dystrophies rétiniennes ou des neuropathies optiques. Ces aspects sont comparables pour la pathologie du segment antérieur avec là aussi des centres spécialisés dédiés en cas de pathologie oculaire isolée ou syndromique de l'enfant. Ce conseil est encadré par des textes législatifs1 et nécessite une réflexion fondée sur la confrontation et la concordance entre les données cliniques, familiales, les résultats et connaissances des gènes candidats et de leurs mutations.
Le criblage d'un ou de quelques gènes a été remplacé par le séquençage haut débit avec l'élaboration de panels évolutifs et dédiés, analysant quelques centaines de gènes (étude d'exons) associés à tels ou tels types de dystrophies et modes de transmission. Le criblage d'un seul gène est de plus en plus rarement pratiqué en dehors de quelques phénotypes (ou tableaux cliniques) précis liés à une anomalie d'un seul gène, tels la choroïdérémie (gène CHM; Rab escort protein1), l'atrophie gyrée (gène OAT ou ornithine aminotransférase), la maladie de Bietti (gène CYP4V2 ou cytochrome P450 family 4-subfamily V member2).
Ces techniques de séquençage à haut débit permettent désormais d'identifier des mutations causales y compris dans les cas dits simplex (pas d'autres apparentés atteints). Le rendement de ces panels est variable, en moyenne supérieur à 50 % des cas. Mais par de tels panels plusieurs mutations candidates dans plusieurs gènes de dystrophies rétiniennes peuvent être identifiées rendant complexe le conseil. Cette problématique de mutations découvertes fortuitement est majeure dans les études de l'exome ou du génome puisque dépassant les gènes associés à des pathologies rétiniennes. Le patient souhaite-t-il être informé de mutations qui pourraient être identifiées dans des gènes associés à des cancers, à des pathologies neurologiques dégénératives, etc.?
Au-delà de la prise en charge du patient et de son information, il restera à aborder les cas particuliers du mineur asymptomatique et de l'information à donner aux apparentés qui pourraient être atteints. Le patient assurera-t-il la diffusion de l'information génétique ou demandera-t-il au médecin prescripteur de donner cette information aux apparentés?
1. Voir en ligne, la Société de génétique ophtalmologique francophone (https://sites.google.com/site/societeophtalmologiegenetique/home) et Legifrance (https://www.legifrance.gouv.fr/affichTexte.do?cidTexte=JORFTEXT000027513617&categorieLien=id)
Petit lexique de génétique
Génotype : caractéristique génétique associée à un individu = la mutation.
Phénotype : caractéristique clinique associée à un individu = la dystrophie.
Autosome : un des chromosomes de 1 à 22 distinct des chromosomes sexuels X et Y.
Gène : séquence d’acide désoxyribonucléique (ADN) contenant l’information génétique nécessaire pour la synthèse d’une ou plusieurs protéines.
Allèle : copie d’un gène. Deux copies pour un individu : une copie héritée de la mère, une copie héritée du père.
Pénétrance : capacité d’un variant pathogène de résulter ou non dans un phénotype donné = porteur asymptomatique.
Expressivité : caractéristiques cliniques de l’expression de la pathologie dans son degré de sévérité. Exemple : cas de maculopathie ou de rétinite pigmentaire pour une même mutation dans une famille.
Variant : un variant pathogène est une mutation, un variant non pathogène est une variation du code ADN sans traduction clinique.
Pathologie dominante : un seul variant pathogène porté par une des deux copies parentales (mutation hétérozygote) est suffisant pour déclencher la pathologie.
Pathologie récessive : deux variants pathogènes, l’un sur l’allèle maternel et l’autre sur l’allèle paternel, sont requis pour déclencher la pathologie (mutations). On distingue des patients hétérozygotes composites lorsque les deux allèles hérités ne portent pas la même mutation et des patients homozygotes lorsqu’ils portent la même (consanguinité).
L'étude génétique est dictée par le diagnostic clinique et l'hypothèse du mode de transmission d'après l'arbre généalogique.
À l'interrogatoire, tout signe, ou pathologie, extra-oculaire est recherché, l'atteinte est soit isolée soit syndromique. Puis, l'examen ophtalmologique :
- – reprend les signes fonctionnels : baisse d'acuité visuelle, héméralopie, photophobie, vision des couleurs, champ visuel isoptère périphérique et zone centrale;
- – et inclut :
- – une imagerie multimodale : clichés en couleurs, autofluorescence et OCT spectral ;
- – des explorations électrophysiologiques : électrorétinogramme (ERG) grand champ systématique, électro-oculogramme (EOG) si dépôts ou pathologies mal étiquetées de l’épithélium pigmentaire, potentiels évoqués visuels (PEV) et ERG multifocal pour différencier l’implication de la macula ou du nerf optique.
Au décours de l'examen, la dystrophie rétinienne est alors classée en :
- – maculopathie;
- – dystrophie des cônes;
- – rétinite pigmentaire ou autres rétinopathies (prédominance de l'atteinte des bâtonnets à l'ERG);
- – dystrophies de type cônes-bâtonnets (prédominance de l'atteinte des cônes à l'ERG) (Tableau 28-1).
L'arbre généalogique doit être minutieux, inclure tous les apparentés, les pathologies oculaires et leurs signes fonctionnels, les pathologies associées, les causes et l'âge des décès, les enfants décédés en bas âge. Les codes utilisés pour la réalisation de l'arbre sont présentés dans la figure 28-16.
Les sujets apparentés et leurs documents ophtalmologiques sont à examiner dans la mesure du possible. En effet, le phénotype ou tableau clinique peut être plus évocateur, car moins évolué chez un apparenté plus jeune (fig. 28-17 et 28-17 ). Dans les cas isolés (cas simplex), il est préférable d'examiner les parents afin de s'assurer qu'aucun d'entre eux ne soit porteur asymptomatique ou atteint (rétinite pigmentaire à pénétrance ou expressivité variable, maladie de Best et porteur asymptomatique). Dans les cas de garçons, la mère est examinée à la recherche de signes discrets notés chez les conductrices (fig. 28-19 ).
Au décours de cette étape, le médecin a une ou plusieurs hypothèses principales :
- – du type de dystrophie : maculopathie versus dystrophie mixte versus neuropathie;
- – du mode de transmission : dominant versus récessif versus lié à l'X.
Les différents modes de transmission sont rappelés dans les eFigures 28-1 à 28-4.
Le médecin prescripteur est soit un médecin généticien, soit un ophtalmologiste connaissant la situation clinique (maladie, prise en charge thérapeutique) et les conséquences familiales, et capable d'en interpréter les résultats. Ce médecin doit travailler en relation avec une équipe de génétique clinique. La liste des centres spécialisés dans ces pathologies ophtalmologiques spécifiques est accessible sur le site Internet de la Société de génétique ophtalmologique francophone2.
Tableau 28-1 -Classification des dystrophies rétiniennes en fonction des signes fonctionnels, de l’imagerie et des explorations électrophysiologiques.
Lecture et interprétations des résultats
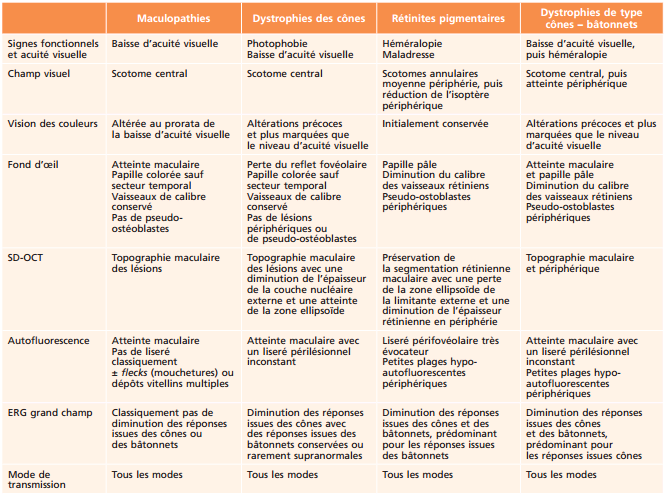
Le médecin prescripteur doit certifier avoir informé le patient ou son représentant légal des caractéristiques de la maladie recherchée, des moyens de la diagnostiquer, des possibilités éventuelles de prévention et de traitement, du stockage du prélèvement, et avoir recueilli son consentement éclairé signé. Il doit conserver le consentement écrit, les doubles de la prescription et de l'attestation, les comptes rendus d'analyses de biologie médicale commentés et signés. Les documents législatifs et des modèles d'attestation et de consentement éclairés sont accessibles sur le site de la Société de génétique ophtalmologique francophone3.
2. Cliquer sur la rubrique Liens utiles à l’adresse Internet suivante : https://sites.google.com/site/societeophtalmologiegenetique/home
3. https://sites.google.com/site/societeophtalmologiegenetique/home
Ces panels sont évolutifs et dédiés à un type de dystrophie et à un mode de transmission. Ils portent sur l'analyse des gènes principaux connus pour être impliqués dans la pathologie concernée. L'absence d'identification de mutations n'exclut ni la pathologie (gènes non connus) ni l'implication de ces gènes testés (mutations introniques).
Par exemple, nous disposons actuellement d'un panel « maculopathie » qui teste les gènes ABCA4, ELOVL4, PRPH2, BEST1 et RS1, d'un panel rétinite pigmentaire non syndromique autosomique dominante, rétinite pigmentaire non syndromique autosomique récessive. Il n'y a pas de panel dédié aux dystrophies des cônes pures.
Le médecin prescripteur n'est plus l'ophtalmologiste. Le conseil génétique est ici rendu par un centre spécialisé ou par un médecin généticien. L'information donnée peut concerner les résultats s'appliquant à la pathologie qui a motivé l'étude génétique mais peut aussi s'étendre à ceux de mutations dans des gènes prédisposant à des cancers si le patient a donné son accord.
Les panels ont révolutionné notre pratique en nous apportant un diagnostic génétique formel dans un nombre de cas croissant y compris dans les cas simplex. Cependant, l'interprétation des résultats est complexe car des variants peuvent être identifiés dans plusieurs gènes associés à des dystrophies rétiniennes. Lequel de ces variants est réellement la mutation causale? Nous prendrons pour exemple la famille F. (fig. 28-20 à 28-22 ). Le sujet index a une rétinite pigmentaire (fig. 28-20 ). C'est la seule personne atteinte dans la famille. Après étude génétique, trois « variants » sont identifiés dans des gènes associés à des rétinites pigmentaires de transmission autosomique dominante. On note une « mutation » référencée dans PRPH2, mutation hétérozygote dans l'exon 2 (c.623C> T, p.G208D), un variant p.Tyr76stop non référencée dans GUCA1A, et une délétion de deux nucléotides dans le gène PRPF31. Le variant c.623C> T dans le gène PRPH2 est également noté chez la mère et la sœur de la patiente. Or, ni l'une ni l'autre n'ont une atteinte oculaire, leurs imageries et leurs ERG sont strictement normaux (fig. 28-21 et 28-22 ). Même si ce variant a été décrit dans la littérature dans un cas d'atrophie aréolaire centrale, il est pathogène pour certains logiciels de prédiction tels PROVEAN™ (protein variant effect analyzer), MutationTaster™, aGVGD™ mais non pathogène pour PolyPhen™. De plus, si l'on tient compte des bases de données, la fréquence de ce variant dans la population serait de 30/1 000 000. Ce seul variant pourrait être dès lors responsable de 1500 cas de rétinites pigmentaires en France. De plus, il n'a jamais été rapporté de variations de pénétrance pour ce gène. Il est ainsi peu probable que ce variant soit une mutation. Passons maintenant au variant dans GUCA1A. Les mutations dans ce gène donnent classiquement une dystrophie de type cônes-bâtonnets, ce qui ne correspond pas au phénotype de rétinite pigmentaire de la patiente. Là encore, le variant dans GUCA1A n'est probablement pas une mutation. Le dernier variant, la délétion de deux nucléotides est probablement la mutation causale de la rétinite pigmentaire dans cette famille, expliquant ainsi les variations de pénétrance.

Fig. 28-16 Modèle pour dessiner l’arbre généalogique imprimé sur un format A3.
Un numéro de famille est donné afin de rattacher tous les apparentés au sujet index atteint.
Ce cas illustre qu'il faut rester prudent dans les cas simplex qui peuvent aussi être liés à une transmission dominante ou liée à l'X chez un garçon. Il apparaît que l'identification d'un variant génétique nécessite une analyse rigoureuse avant de conclure à son rôle certain dans la pathologie. Dans ce contexte, l'enquête génétique nécessite une coopération entre cliniciens et généticiens. Le retour vers le clinicien du résultat génétique permet des corrélations phénotypes/génotypes validées.
Le résultat génétique lié à la pathologie est expliqué au patient si et seulement si ce dernier l'a souhaité et l'a formulé par écrit (consentement pour l'examen stipulant « Je souhaite être informé du résultat oui/non »). L'information donnée doit être claire et appropriée. La consultation est accompagnée d'un compte rendu qui résume à nouveau les conséquences pour l'individu, le mode de transmission et les conséquences familiales, les modalités d'information de la parentèle (patient ou médecin prescripteur) et la copie du résultat du laboratoire de biologie médicale qui a réalisé l'étude génétique.
Le patient peut être orienté vers une consultation de conseil génétique complémentaire, vers un spécialiste de la maladie considérée en particulier dans les formes syndromiques, notamment dans le cas d'un projet parental ou d'une information sur des résultats génétiques sans lien direct avec la pathologie, mais pouvant avoir un impact sur sa santé ou celle d'apparentés; il faut savoir si ces résultats étaient souhaités par le patient (voir plus haut « Consentement »). Un accompagnement psychologique peut être également discuté avec le patient.
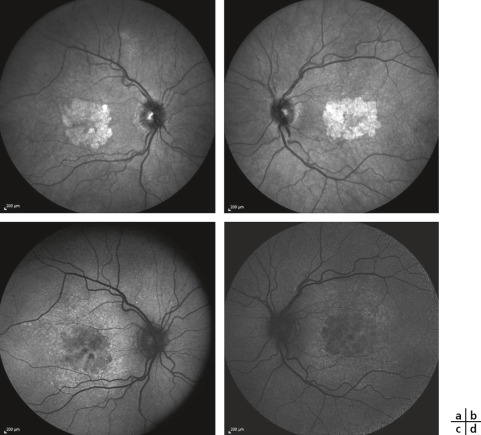
Fig. 28-17 Patient âgé de 66 ans qui consulte pour des difficultés de lecture, de reconnaissance des visages et d’adaptation aux variations de luminosité.
Il est discrètement photophobe. L’acuité est de 6/10 aux deux yeux. Il a un diabète non insulino-dépendant traité depuis 3 ans. Au fond d’oeil (a, b), on note une atrophie choriorétinienne maculaire bilatérale et symétrique. Il n’y a pas de drusen. Sur les clichés en autofluorescence (c, d), la fovéa est respectée en partie expliquant la conservation de l’acuité visuelle. Au-delà de l’atrophie, l’autofluorescence rétinienne est irrégulière avec un petit piqueté hyperautofluorescent. Le diagnostic retenu chez l’index est celui d’une maculopathie héréditaire de transmission autosomique récessive, les parents de l’index n’ayant pas eu de baisse d’acuité visuelle. Sa soeur rapporte également des difficultés visuelles avec les résultats suivants (voir fig. 28-18 ).
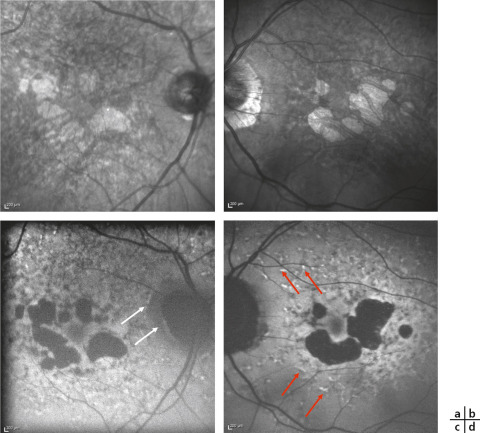
Fig. 28-18 Lasœur du patient de la , âgée de 68 ans, a des signes comparables et là encore une acuité visuelle relativement préservée à 8/10. L'aspect est ici très en faveur d'une maladie de Stargardt de l'adulte avec une périfovéopathie, des taches flavimaculées hyperautofluorescentes prédominantes au pôle postérieur ( : flèches rouges) et un signe très évocateur, l'épargne péripapillaire, qui ne correspond à aucune atrophie péripapillaire à droite ( : flèches blanches). Au total, suite à l'examen de la sœur, le diagnostic est celui d'une périfovéolopathie de Stargardt. Ce diagnostic a été confirmé par l'étude génétique du gène Cette analyse génétique a confirmé la transmission autosomique récessive.
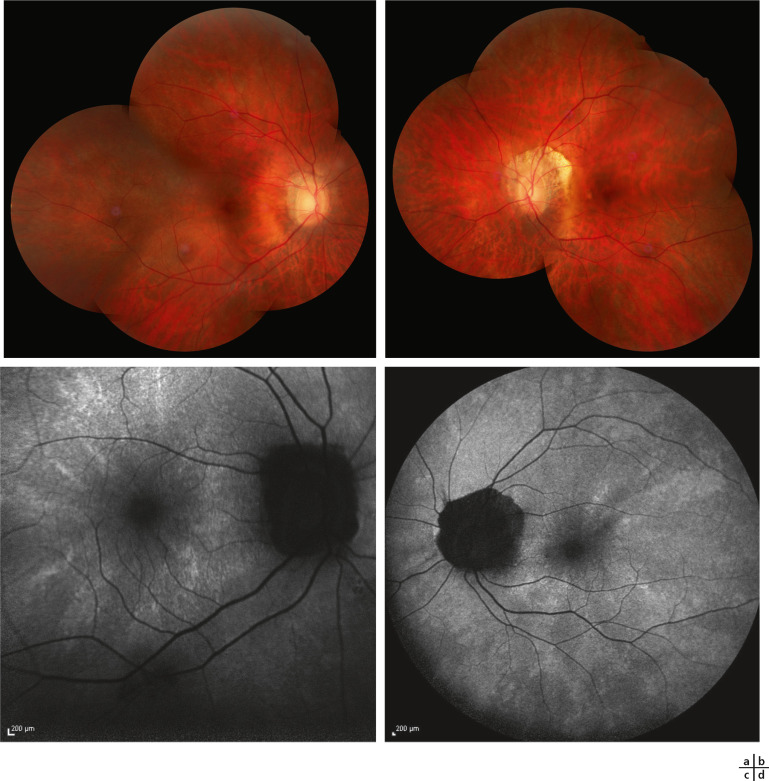
Fig. 28-19 Femme conductrice de rétinite pigmentaire liée à l’X, intérêt des clichés en autofluorescence. La patiente n’a pas d’héméralopie ni de réduction du champ visuel périphérique.
Elle est myope forte (−9 oeil droit et gauche). Sur les clichés en couleurs (a, b), il est retrouvé une choroïdose et un conus myopique. Par contre, l’aspect radiaire et peigné au pôle postérieur noté sur les clichés en autofluorescence (c, d) signe que la patiente est conductrice.
D'après la loi, dans le consentement pour l'examen génétique, le patient précise s'il souhaite assurer lui-même la diffusion de l'information génétique aux membres de sa famille ou s'il autorise le médecin prescripteur à cette diffusion.
Les apparentés à risque (en fonction du mode de transmission) symptomatiques majeurs ou mineurs peuvent être examinés et testés avec leur accord (consentement pour l'examen génétique à signer par les parents ou le tuteur pour un mineur). Là encore, l'apparenté précise s'il souhaite ou non être informé du résultat génétique.
Le texte de loi pour les pathologies sans traitement stipule que « la prescription d'un examen génétique susceptible d'annoncer la survenue d'une maladie grave chez un sujet asymptomatique, sans option de traitement ou de prévention ou avec des possibilités de traitement et de prévention limitées doit être effectuée dans le cadre d'une consultation individuelle par un médecin exerçant au sein d'une équipe pluridisciplinaire de prise en charge des patients asymptomatiques en raison des conséquences potentiellement délétères d'une information incomplète ou mal comprise. Cette équipe doit valider la bonne préparation de la personne à la réalisation du test ».
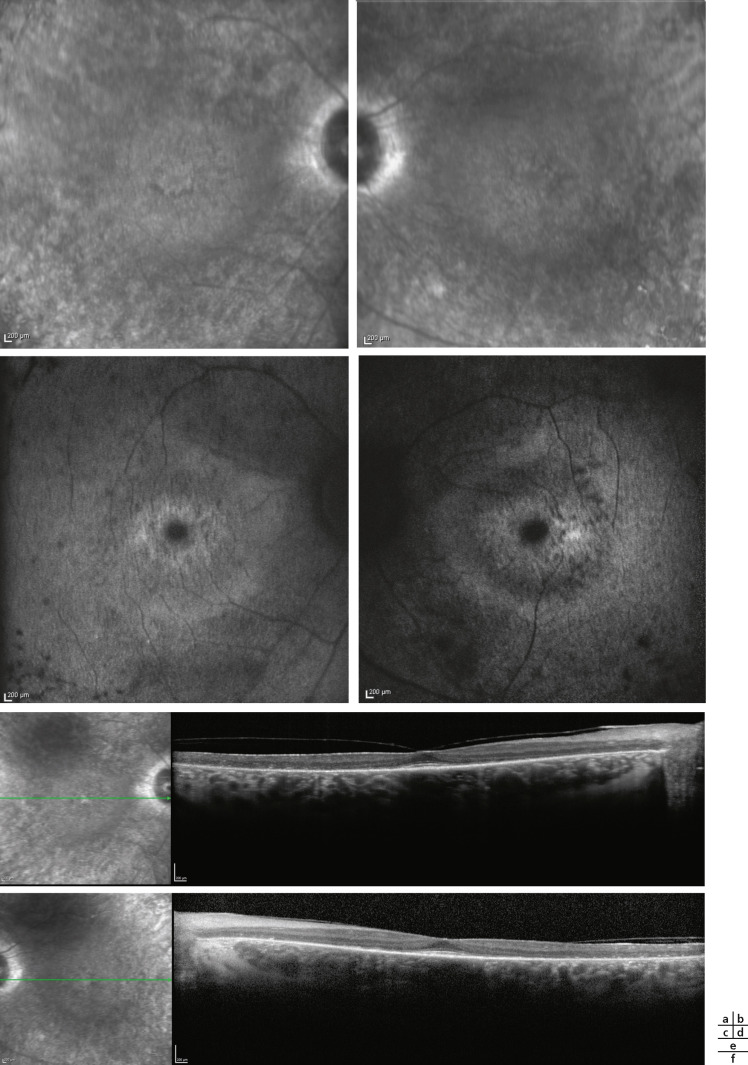
Fig. 28-20 Difficultés du conseil génétique : le faux cas simplex avec une mutation causale dans un gène d’épissage et ses variations de pénétrance.
Il s’agit d’une jeune femme vue initialement à l’âge de 15 ans en 2001 avec une héméralopie, une acuité visuelle de 10/10 aux deux yeux et un ERG altéré avec des réponses issues des cônes et des bâtonnets non discernables. En 2015, l’acuité visuelle est toujours de 10/10 aux deux yeux, mais le champ visuel est tubulaire sur les 20° centraux avec des îlots temporaux. L’imagerie confirme le diagnostic de rétinite pigmentaire avec une réduction du calibre des vaisseaux rétiniens (a, b), un liseré périfovéolaire en autofluorescence (c, d) correspondant à la zone de transition à l’OCT (e, f), et de multiples petites plages hypoautofluorescentes en moyenne périphérie. De principe, la mère et la soeur ont été examinées (voir fig. 28-20 et 28-21 ).
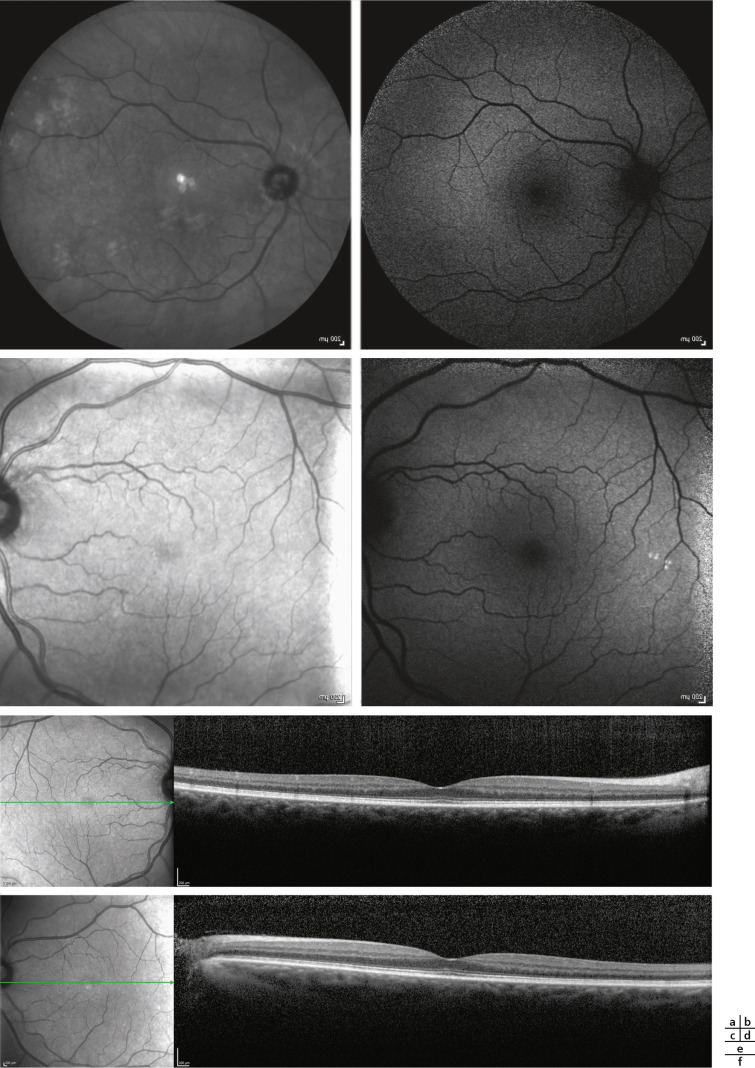
Fig. 28-21 Imagerie multimodale de la mère.
a, b. Clichés en infrarouge. c, d. Clichés en autofluorescence. e, f. SD-OCT oeil droit (e) et oeil gauche (f). La mère est asymptomatique et n’a aucune anomalie rétinienne. Elle est porteuse des variations dans PRPH2 et dans PRPF31. Ces variations ont été transmises à ces deux filles (cas index, voir fig. 28-19 ).Imagerie multimodale de la soeur de l’index.
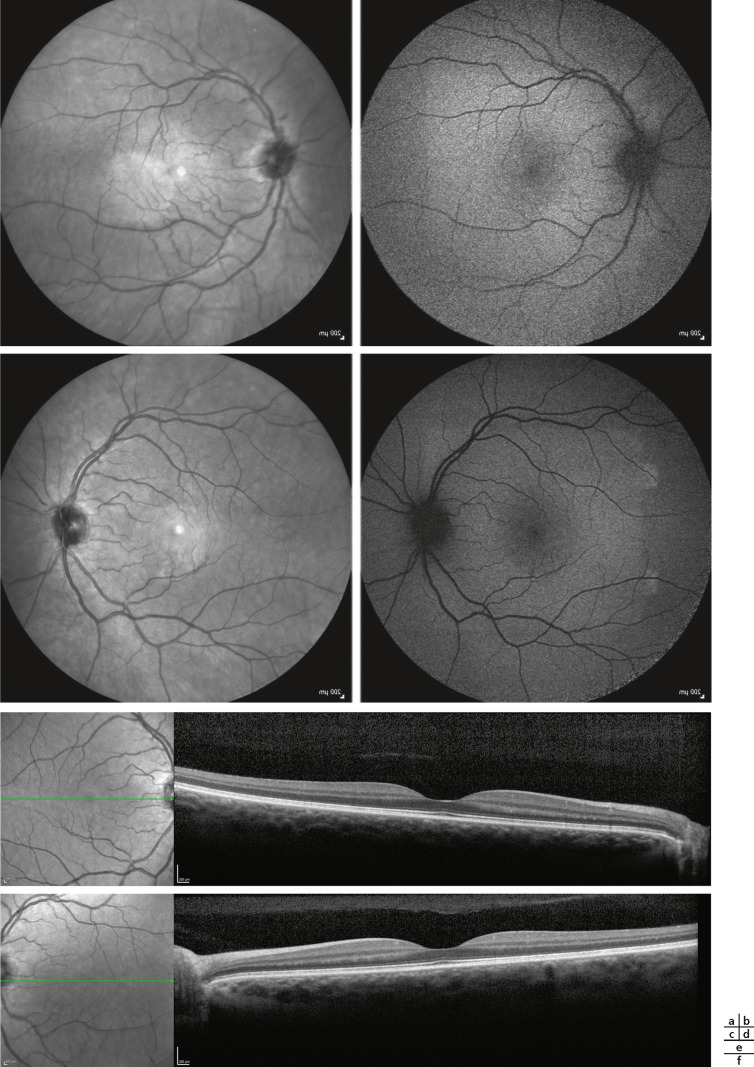
a, b. Clichés en infrarouge. c, d. Clichés en autofluorescence. e, f. SD-OCT oeil droit (e) et oeil gauche (f). La soeur est asymptomatique et n’a aucune anomalie rétinienne. Elle est également porteuse des variations maternelles dans PRPH2 et dans PRPF31. Compte tenu de l’absence d’atteinte de la mère et de la soeur, la variation causale de la pathologie de l’index est celle notée dans PRPF31 qui est un gène d’épissage.
Le conseil génétique dans le cadre d'un projet parental incombe à des centres spécialisés en conseil génétique mais l'ophtalmologiste a un rôle clé initialement par le diagnostic précis de l'atteinte oculaire et les modes de transmission possibles. L'ophtalmologiste peut aussi être sollicité dans un deuxième temps pour préciser la sévérité de l'atteinte, les variations intrafamiliales de sévérité ou de pénétrance.
Le projet parental reste une démarche lourde et longue pour les parents avec de possibles refus de grossesse médicalement assistée pour des pathologies oculaires de gravité variable ne mettant pas en jeu le pronostic vital, exception faite des amauroses congénitales de Leber ou des rétinites pigmentaires syndromiques.

Fig. 28-23 Interactions entre ophtalmologistes et généticiens.
La place de l'ophtalmologiste est essentielle dans ces maladies oculaires génétiques, car le généticien n'en a pas la connaissance clinique. Mais de même, l'ophtalmologiste n'a pas à lui seul les connaissances génétiques. Le conseil génétique reste difficile car nous entrons dans une famille, son histoire, ses non-dits, ses ruptures, la culpabilité d'avoir transmis la pathologie. Il est également compliqué car le mode de transmission peut paraître évident sur l'arbre généalogique mais ce serait sans compter sur les variations de pénétrance (notamment dans les cas de mutations causales dans des gènes d'épissage), les variations de sévérité, les mutations de novo (où le patient atteint est le premier maillon d'une transmission dominante), une consanguinité méconnue par isolat géographique et des défauts de paternité.
La figure 28-23 résume les interactions entre ophtalmologistes et généticiens.
Pour en savoir plus
Attestation du médecin prescripteur. Document du CHU de Montpellier. En ligne (rubrique Liens utiles) : https://sites.google.com/site/societeophtalmologiegenetique/home
Conseil de l’Europe. Les tests génétiques à des fins médicales.2012.
Modèle de consentement de patient. Document du CHU de Montpellier. En ligne (rubrique Liens utiles) : https://sites.google.com/site/societeophtalmologiegenetique/home
Modes de transmission. Document du CHU de Montpellier. En ligne (rubrique Liens utiles) : https://sites.google.com/site/societeophtalmologiegenetique/home
Orphanet, le portail des maladies rares. En ligne : www.orpha.net
Société de génétique ophtalmologique francophone. Liste des centres de référence et de compétence nationaux. En ligne (rubrique Liens utiles) : https://sites.google.com/site/societeophtalmologiegenetique/home
A. Péchereau
La notion d'emmétropisation a été abordée par G. Glergeau dans le Rapport de la Société Française d'Ophtalmologie de 2013 [3] et par le même auteur de façon encore plus détaillée dans la deuxième édition de son livre consacré à La réfraction de l'enfant ([5], p. 264-294).
Dans ce chapitre, nous ne parlerons pas ou peu de l'évolution de l'astigmatisme qui est considéré comme un élément relativement stable sur la longue durée (même s'il ne l'est pas sur la très longue durée et durant les premières années de vie). De même et sauf mention contraire, notre propos considérera que l'accommodation du sujet est nulle naturellement (âge) ou grâce à l'utilisation d'un cycloplégique fort.
L'emmétropisation serait un phénomène dynamique d'adaptation de la puissance optique de l'œil entraînant un pourcentage « élevé » (une sur-représentation) d'« emmétrope » (en fait de faible hypermétrope) dans la population.
Ce phénomène fait l'objet de nombreuses études puisque l'on dénombre plus de 340 références bibliographiques à la fin de l'année 2016.
Le phénomène dit d'emmétropisation recouvre deux phénomènes distincts :
- – une distribution non gaussienne des amétropies. Cette idée est fondée sur le constat d'une sur-représention des hypermétropies faibles à modérées dans la population générale des adultes par rapport à une courbe de distribution normale;
- – une moyenne des réfractions voisines de zéro. Cette idée est fondée sur le constat que les enfants naissent plutôt hypermétropes et que la réfraction moyenne de leur population évoluerait statistiquement vers une « emmétropie » à la fin de l'enfance ou de l'adolescence.
Le concept d'emmétropisation est né au début du xx siècle. Il est attribué par certains à : Straub (1909) [27] ou Steiger (1913) [26]. Pour ces auteurs, ce concept correspond plutôt à la deuxième définition, c'est-à-dire une moyenne des réfractions voisines de zéro.
Avec Brown, on peut être extrêmement critique : « En 1913, Steiger a étudié des centaines d'écoliers suisses âgés entre 6 et 11 ans. Il a développé à partir de là sa théorie biologique de l'hérédité des états de réfraction et des changements qui ont lieu dans les yeux des enfants en pleine croissance. La réfraction ayant été déterminée par la méthode “manifeste”, sans cycloplégie, ses conclusions sont susceptibles d'être critiquées » [2]. Malgré ce constat, ce concept a eu une postérité abondante.
Le système optique de l'œil est composé de trois éléments principaux (fig. 28-24 ) :
- – la cornée. Elle représente le principal facteur réfractif de l'œil, soit les deux tiers de la puissance optique de l'œil. Avec les outils actuels, sa puissance peut être calculée avec précision;
- – le cristallin. Celui-ci comprend deux éléments :
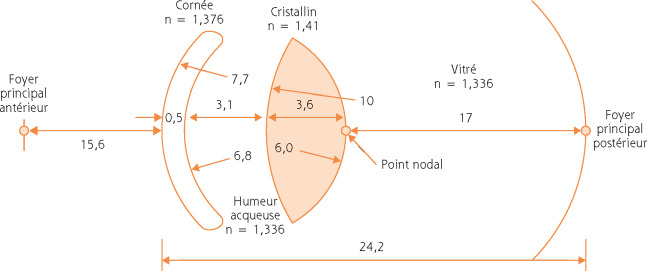
Fig. 28-24 Les différents composants de la réfraction d'un œil.
- – le cristallin. Celui-ci comprend deux éléments :
- – un élément fixe. Il correspond à la puissance de base du cristallin. Nous ne savons pas pour l’instant la calculer de manière directe. Les calculs de puissance de l’implant sont des méthodes indirectes le plus souvent fondées sur des régressions linéaires ;
- – un élément variable. Celui-ci correspond à l’accommodation. Malgré l’emploi de cycloplégiques forts, nous n’avons pas les moyens de contrôler avec précision l’état accommodatif. Plus le sujet est jeune, plus ce problème est important. Il faut attendre une presbytie bien avancée (≥ 50 ans) pour que l’élément accommodatif puisse être considéré comme négligeable. La cornée et le cristallin font un ensemble de deux lentilles épaisses (pour plus de précision voir [22]) et qui correspond à la puissance dioptrique de l’oeil ;
- – la longueur axiale. Elle détermine la position du capteur rétinien (fig. 28-25).
L'inadéquation entre la puissance optique de l'œil (celui-ci étant au repos, c'est-à-dire n'accommodant pas) et la longueur axiale est l'explication de toutes les amétropies sphériques. Il est important de distinguer l'origine de cette inadéquation. Elle peut être de trois types :
- – les amétropies axiales. La longueur de l'œil est trop courte (hypermétropie) ou trop longue (myopie) pour une puissance optique « normale »;
- – les amétropies de puissance. La puissance optique est trop forte (hypermétropie) ou trop faible (myopie) pour une longueur de l'œil « normale »;
- – les amétropies mixtes. Naturellement, chaque œil va avoir sa propre combinaison et le nombre des possibilités est infini.
fig. 28-25 Trois types d'amétropies : les amétropies axiales, les amétropies de puissance et les amétropies mixtes.
La cornée est l'élément optique le plus puissant de l'œil. Mais, contrairement à ce que l'on pourrait penser, pour O. Touzeau, « la courbure de la cornée ne joue pas un rôle déterminant dans les amétropies sphériques… Les différences de courbure sont faibles selon le groupe réfractif et la courbure cornéenne moyenne n'est pas corrélée à l'équivalent sphérique. La toricité de la cornée explique l'essentiel de l'astigmatisme réfractif et la symétrie en miroir des axes (énantiomorphisme). L'astigmatisme réfractif est d'autant plus proche de l'astigmatisme cornéen que le cylindre est élevé. L'épaisseur de la cornée, son diamètre, son asphéricité n'ont pas d'influence sur la réfraction. Bien que la cornée soit responsable des deux tiers du pouvoir réfractif de l'œil, en dehors de la toricité, la géométrie de la cornée a finalement peu d'influence sur la réfraction » [29].
Les études recherchant une corrélation entre la puissance de la cornée et l'amétropie ont bien confirmé ces affirmations : il n'y a pas de corrélation ou elle est très faible (fig. 28-26 et 28-27 ).
La longueur axiale a un rôle majeur dans l'amétropie comme nous pouvons le voir (fig. 28-29 et 28-29). L'examen de ces deux figures montre :
- – une excellente corrélation entre longueur axiale et réfraction;
- – une certaine dispersion dans la relation amétropie-longueur axiale, en particulier dans les amétropies fortes (fig. 28-29 ).
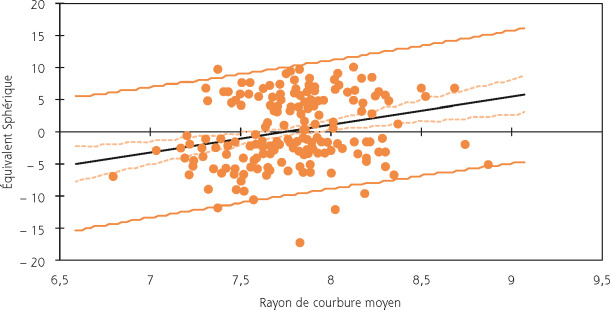
Fig. 28-26 Corrélation entre l'équivalent sphérique cycloplégié et la puissance de la cornée (équivalent sphérique cycloplégié = –33,36 +4,31*Rayon de courbure moyen, OD, nb : 213, R : 0,067, t : 3,897, p : 0) [7].
C'est le rapport longueur axiale (AL pour axial length) par rayon de courbure (CR pour corneal radius). Il est utilisé depuis de nombreuses années [12, 20]. Un chiffre supérieur à 3 est souvent avancé pour indiquer l'existence d'une myopie. Les coefficients de détermination entre ce paramètre et l'équivalent sphérique sont très souvent supérieurs à ceux entre l'équivalent sphérique et la longueur axiale.
Dans les amétropies et bien qu'étant l'élément le plus puissant, la variation du rayon de courbure de la cornée n'explique que 7 % de la variation des amétropies.
En revanche, la variation de la longueur axiale explique à elle seule 80 % de la variation de l'amétropie, ce qui est considérable. C'est pourquoi le problème de la définition des amétropies sphériques se pose.
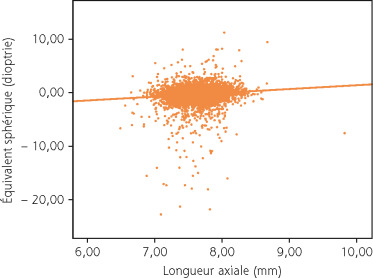
Fig. 28-27 Absence de corrélation entre la puissance de la cornée et l'équivalent sphérique [13].
Les amétropies sont une inadéquation entre la puissance optique (l'œil étant au repos, c'est-à-dire n'accommodant pas) et la longueur axiale. Cette inadéquation peut provenir de la cornée, du cristallin (mais les moyens objectifs de calculer la puissance du cristallin n'existant pas, nous l'éliminerons comme cause, bien qu'elle explique sÛrement certaines amétropies) et de la longueur axiale. De ce fait deux types de définitions peuvent être proposés.
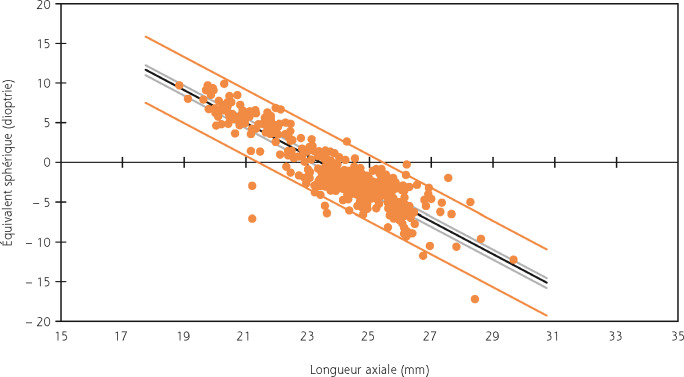
Fig. 28-28 Corrélation entre l'équivalent sphérique cycloplégié et la longueur axiale (équivalent sphérique cycloplégié = 48,17 –2,05*longueur axiale, OD, nb : 340, R : 0,795, t : –36,096, p < 0,0001) [7].
On distingue deux types d'amétropies sphériques en fonction du type de puissance du verre correcteur.
L'œil n'accommodant pas et fixant un objet à l'infini, l'image se forme en arrière de la rétine. La correction se fait par un verre positif. C'est ce qui est appelé l'hypermétropie.
L'œil n'accommodant pas et fixant un objet à l'infini, l'image se forme en avant de la rétine. La correction se fait par un verre négatif. C'est ce qui est appelé la myopie.
Considérant qu'un œil normal a une longueur comprise entre 21,54 mm et 24,3 mm [21, 28], on peut également définir les yeux par la biométrie.
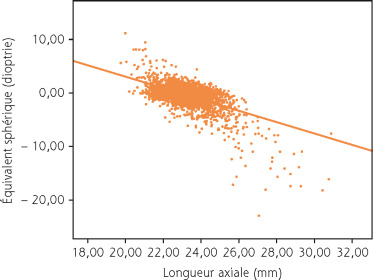
Fig. 28-29 Absence de corrélation entre la puissance de la cornée et l'équivalent sphérique [13].
Ce sont les yeux dont la longueur axiale est inférieure à 21,54 mm.
Ce sont les yeux dont la longueur axiale est supérieure à 24,3 mm, une longueur axiale supérieure à 26 mm définissant la myopie maladie.
L'étude fine des biométries montre que, pour une emmétropie définie comme allant de –0,5 D à +0,5 D, la longueur axiale peut aller de 22,25 mm à 26,20 mm [7]. Dans ce dernier cas (avec longueur axiale de 26,20 mm pour un équivalent sphérique cycloplégié de –0,25 D), le sujet se présente comme un emmétrope alors qu'il a une longueur axiale de myopie maladie. Ces observations nous renvoient à la notion de normalité [11] que nous avons déjà abordée au chapitre 2.1.
La réponse est clairement non. Le lecteur pourra trouver à la référence [21] quelques exemples cliniques illustrant ce propos.
Il existe des myopes ayant des yeux courts du fait d'un très petit rayon de courbure cornéenne (forte puissance) et des hypermétropes ayant des yeux longs du fait d'un très grand rayon de courbure cornéenne (faible puissance). C'est ce qui explique l'absence de corrélation entre l'équivalent sphérique et le rayon de courbure moyen.
Ces deux groupes n'étant pas superposables, il est indispensable d'aborder ces deux notions de front et de les étudier simultanément.
- – L'amétropie optique. Elle est une conséquence. Elle est difficile à retenir de façon isolée car elle correspond, pour chaque sujet étudié, à une alchimie particulière de trois paramètres ayant des possibilités évolutives très différentes : la puissance de la cornée, la puissance du cristallin et la longueur axiale.
- – La puissance de la cornée. Elle peut être calculée de façon objective. Nous pourrions retenir ce critère mais nous avons vu qu'il n'est que peu corrélé avec l'amétropie.
- – La puissance du cristallin. Nous n'avons pas de moyen objectif de calculer sa puissance. Les méthodes utilisées pour calculer les implants sont des méthodes indicatives et/ou indirectes fondées pour la plupart sur des outils de régression statistique. De plus, chez le sujet jeune, nous ne contrôlons pas ou mal l'accommodation.
- – La longueur axiale. C'est le paramètre de la réfraction qui explique la majeure partie du phénomène amétropique et de son évolution.
- – L'amétropie optique. Elle est une conséquence. Elle est difficile à retenir de façon isolée car elle correspond, pour chaque sujet étudié, à une alchimie particulière de trois paramètres ayant des possibilités évolutives très différentes : la puissance de la cornée, la puissance du cristallin et la longueur axiale.
- – La puissance de la cornée. Elle peut être calculée de façon objective. Nous pourrions retenir ce critère mais nous avons vu qu'il n'est que peu corrélé avec l'amétropie.
- – La puissance du cristallin. Nous n'avons pas de moyen objectif de calculer sa puissance. Les méthodes utilisées pour calculer les implants sont des méthodes indicatives et/ou indirectes fondées pour la plupart sur des outils de régression statistique. De plus, chez le sujet jeune, nous ne contrôlons pas ou mal l'accommodation.
- – La longueur axiale. C'est le paramètre de la réfraction qui explique la majeure partie du phénomène amétropique et de son évolution.
L'amétropie optique seule n'est pas un paramètre pertinent pour étudier la variation évolutive de l'adéquation ou de l'inadéquation de la puissance de l'œil à sa longueur axiale. C'est un paramètre intéressant mais il s'agit plus de la conséquence que de la cause.
Le paramètre clé en fonction des données actuelles est la longueur axiale; les études à venir devraient être organisées en fonction de ce paramètre.
Toute étude portant sur l'évolution des amétropies (phénomène d'emmétropisation ou autre) devra étudier de façon conjointe la réfraction cycloplégiée et la longueur axiale. Le hasard statistique peut faire que deux populations étudiées ne sont pas comparables sur ces points.
De plus, les biométries systématiques chez les enfants de même âge et présentant une amétropie évolutive montrent que plus un œil est court, moins il s'allonge, et que plus il est long, plus il s'allonge [16].
Tout cela confirme combien la longueur axiale est un facteur essentiel dans l'étude de l'évolution des amétropies.
Naturellement, ce serait faire preuve d'anachronisme que de retenir ces critères pour les publications les plus anciennes, mais cela reste un argument critique puissant dans leur évaluation.
Avant de poursuivre, il est nécessaire de revenir sur les études classiques portant sur l'emmétropisation. Trois sont particulièrement remarquables : l'étude de Brown (1938) [2]; l'étude de Slataper (1950) [24]; l'étude de Sorsby (1960) [25].
Ces trois études ont donné leurs lettres de noblesse à ce concept. Les deux premières peuvent être regroupées car elles étudient le même paramètre : l'évolution de la réfraction au cours du temps chez une population d'enfants. Elles comprennent un nombre impressionnant de patients. La troisième aborde le problème de la « leptokurticité4 » de la réfraction (sur-représentation d'hypermétropies faibles ou moyennes) dans une population normale d'adultes jeunes.
4. La leptokurticité « désigne une analyse technique mathématique utilisant une loi de distribution probabiliste particulière c’est-à-dire non normale. La grande majorité des événements sont concentrés autour de la moyenne mais certains événements se produisent jusqu’à trois écarts-type de part et d’autre de la moyenne. C’est la différence avec la distribution normale, où l’on n’observe plus d’événements dès que l’on s'éloigne de la moyenne » (https://fr.wikipedia.org/wiki/Leptokurticit%C3%A9).
Présenté en 1938, ce travail [2] comprend 8820 mesures chez 1203 patients strabiques et non strabiques de la naissance à 35 ans au moment de l'inclusion. L'atropine a été utilisée comme cycloplégique. La réfraction a été déterminée par la skiascopie.
« Mes conclusions (fig. 28-30 ) sont les suivantes :
- – l'hypermétropie augmente jusqu'à la fin de la septième année;
- – de 8 à 13 ans, la myopie augmente et l'hypermétropie diminue rapidement;
- – de 14 à 20 ans, cette évolution continue chaque année, mais à une vitesse plus lente (la moitié de la période précédente). La puberté tend éventuellement à provoquer une “emmétropisation”;
- – l'augmentation de la myopie après l'âge de 20 ans est pratiquement négligeable. »
Ce travail était tout à fait remarquable, d'autant plus que la composante longitudinale est forte avec une moyenne de 8 skiascopies par patient, ce qui est à souligner. Cependant, il amène à un certain nombre de critiques :
- – le nombre de cas inclus par année est faible : 35;
- – l'inhomogénéité des inclusions n'est pas précisée;
- – l'analyse a porté sur l'amétropie moyenne, ne permettant pas de mettre en évidence des phénomènes particuliers;
- – il s'agit d'une population sélectionnée et non d'une population générale, puisqu'il s'agit de la clientèle d'un ophtalmologiste.
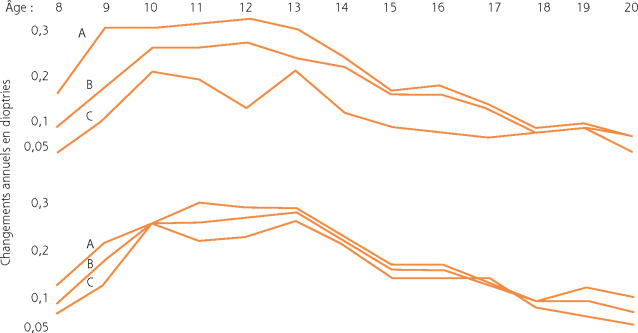
Présenté en 1950, ce travail [24] comprend 35 570 mesures; 14 868 patients ont eu une seule mesure et 20 702 mesures ont été faites plusieurs fois chez un même patient. Le nombre de patients ayant eu plusieurs mesures n'est pas précisé. L'homatropine a été utilisée comme cycloplégique. La réfraction a été déterminée par la skiascopie.
Synthétisées sur la figure 28-31, les conclusions ont été exprimées de cette façon :
- – « la réfraction statique moyenne à la naissance est +2,32 D;
- – l'hypermétropie augmente de la petite enfance à l'âge de 7 ans de +1,617 D;
- – la myopie axiale des jeunes (de 8 à 30 ans) augmente de –3,327 D;
- – le changement hypermétrope pour l'âge moyen (31 ans à 64 ans) est de + 1,362 D;
- – le changement myopique sénile (de 65 à 87 ans) est de –2,367 D. »
Bien que le nombre d'examens soit considérable, ce travail souffre de beaucoup de biais méthodologiques :
- – le cycloplégique utilisé a été l'homatropine qui est un cycloplégique médiocre;
- – la population recrutée est celle d'un ophtalmologiste et non représentative de la population générale;
- – les myopies ont été incluses dans des proportions indéterminées;
- – bien que les résultats soient présentés de façon longitudinale, il s'agit d'études transversales juxtaposées sans contrôle du caractère comparable des populations. Le caractère évolutif de la courbe est-il le reflet d'une véritable évolution ou d'un hasard statistique dÛ aux caractéristiques particulières des diverses populations étudiées? Ce point ne peut pas être tranché.
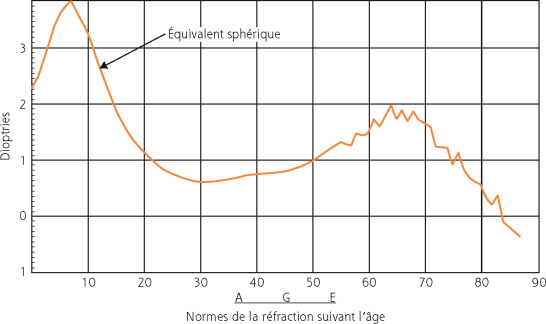
Fig. 28-31 Évolution de l’amétropie selon Slataper [24].
La figure a été simplifiée par l’auteur. Elle montre l’évolution de l’équivalent sphérique.
Ces deux travaux ont été un élément essentiel dans l'acceptation par la communauté ophtalmologique d'une des deux facettes du concept d'emmétropisation. Nous venons de voir que le problème reste entier.
La sur-représentation des hypermétropies faible ou moyenne dans une population normale d'adultes jeunes fait suite à la publication de Sorsby [25].
Cette étude a suivi le protocole suivant :
- – cycloplégique : cyclopentolate;
- – méthode : skiascopie;
- – réfraction étudiée : l'équivalent sphérique;
- – population : militaires;
- – nombre de patients : 1033 patients; sur une base de 1058 sujets (de 17 à 27 ans), 39 ont été éliminés pour diverses raisons (sujets éliminés du service national parce qu'ils avaient une erreur réfractive) et 16 ont été rajouté (13 myopes et 3 hypermétropes).
Les résultats sont synthétisés sur cette célèbre courbe (fig. 28-32 ) représentée de nombreuses fois dans la littérature. Le phénomène de sur-représentation des hypermétropies faible ou moyenne semble évident.
Sur le plan de la conduite des examens, l'approche est rigoureuse et n'est pas soumise à discussion.
En revanche, la population étudiée pose un problème : « comme elle se compose d'hommes reconnus aptes pour le service national, cet échantillon doit être considéré comme pondéré dans le sens des membres les plus en forme de la population masculine. Il existe certaines preuves [23] que les recrues rejetées pour des raisons autres que la vision peuvent aussi avoir une vision généralement plus pauvre que ceux qui n'ont été pas rejetés. L'ampleur de cette corrélation n'est pas connue avec précision, et il n'y a pas de base statistique pour ajuster notre matériel. Tout échantillon sur la base de l'Armée est donc probablement sélectionné dans le sens d'une meilleure vision [25] ».
En d'autres termes, cette étude n'a pas étudié la totalité des conscrits mais les sujets retenus pour leur service militaire. Si le biais de recrutement ne peut pas être affirmé sur les seules données à notre disposition, il peut être fortement suspecté (d'ailleurs, Sorsby lui-même en convient).
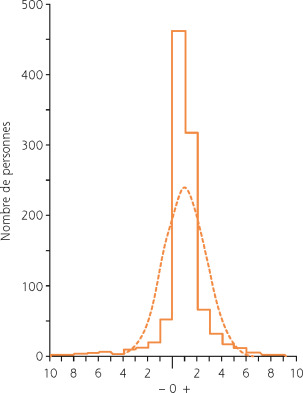
Fig. 28-32 Répartition des amétropies sphériques d’après Sorsby [25].
Le phénomène de sur-représentation des hypermétropies faible ou moyenne est net. Cette courbe est d’ailleurs très fréquente dans la littérature mais est-elle représentative de la population générale (voir texte).
Comme nous venons de le voir, la notion d'emmétropisation est un concept ancien. Chez l'homme, des travaux entre 1938 et I960 ont semblé lui donner ses lettres de noblesse. Nous avons vu que les trois publications princeps comportaient des biais méthodologiques importants.
Depuis, ce concept d'emmétropisation a donné une avalanche de publications tant sur le plan de la recherche fondamentale que sur le plan des données épidémiologiques.
Cependant, la lecture de cette littérature, sans remettre en question le concept, amène à poser toujours les mêmes questions et se heurte toujours au même problème : la représentativité des populations étudiées (le nombre de sujets étudiés ne résout pas la question). On souhaiterait des enquêtes épidémiologiques sur des populations globales (la totalité d'une classe d'âge d'une grande ville, la totalité des enfants d'une ville moyenne suivie pendant 20 ans, etc.) qui utiliseraient systématiquement un cycloplégique fort et les paramètres biométriques de la réfraction. Énoncer ces conditions suffit à comprendre qu'il est très difficile voire impossible de les remplir aujourd'hui.
Pour l'étude du phénomène d'emmétropisation, le phénomène myopique pose un problème difficile. En effet, d'un point de vue mathématique, la myopisation et son évolution entraînent automatiquement une emmétropisation d'une population infantile que l'on suit. Ce problème est un vrai casse-tête qui n'a pas trouvé de solutions satisfaisantes.
Pour certains auteurs [10, 19], la myopie pourrait être la conséquence d'un dérèglement du processus d'emmétropisation.
Nous n’avons pas la place pour faire une analyse complète de la littérature actuelle et, bien que nombre de ces études présentent des limites méthodologiques, cette citation d’Ojaimi [20] nous semble le mieux refléter la situation des connaissances actuelles : « Malgré les limites liées aux méthodes et aux échantillons, le travail de Larsen [18] chez les enfants danois, de Fledelius [8, 9], l’Orinda Longitudinal Study on Myopia [32] avec son extension dans l’étude CLEERE [31], et le COMET [13] ont fourni des données utiles à des fins de comparaison.
>Il existe de grands ensembles de données provenant de populations plus âgées d'origine européenne [1] et d'Asie orientale [30] qui fournissent des données significatives sur la réfraction et sur d'autres affections oculaires, bien que la collecte de la biométrie oculaire soit moins complète. Une méta-analyse des données sur celles d'origine européenne a récemment été publiée et montre, entre autres, des différences régionales significatives entre les erreurs de réfraction en Europe et en Amérique du Nord d'une part et en Australie d'autre part [17]. En particulier, l'Australie présente des taux de prévalence spécifiques plus faibles pour les erreurs réfractives que l'Europe occidentale et les États-Unis.
Malgré les limites des données existantes, en grande partie sur la base des données sur les populations adultes et sur les premiers travaux sur les enfants [6] une relation entre l'erreur de réfraction et la biométrie oculaire est apparue. La plupart des paramètres biométriques oculaires sont normalement distribués, en cohérence avec le fait que leur développement est contrôlé par un grand nombre de facteurs indépendants, à la fois génétiques et environnementaux. Cependant, l'équivalent sphérique montre une distribution maximale et indique qu'elle est sous une certaine forme de régulation active. »
En d’autres termes, on peut résumer la situation actuelle : il existe bien une modification statistique de type « leptokurtique » (surréprésentation d’hypermétropies faibles ou moyennes ; en mathématique leptokurtique = « se dit d’une distribution de probabilités dont la cloche est plus pointue que celle de la loi gaussienne, avec des queues plus importantes5 »), mais cette sur-représentation est beaucoup plus discrète que celle notée dans l’étude de Sorsby.
5. Source : https://fr.wiktionary.org/wiki/leptokurtique.
Les données expérimentales sont nombreuses, variées et parfois contradictoires. De très nombreuses hypothèses ont été avancées et de nombreux mécanismes physiopathologiques ont été proposés. Faire une revue complète de la littérature occuperait la totalité de ce rapport et, aujourd'hui, n'aboutirait pas à des conséquences pratiques.
Ce sont les raisons qui nous ont amené à ne pas aborder cette partie du problème dans ce texte. Nous laissons au lecteur passionné par ce sujet le plaisir de la lecture d'une littérature fort abondante.
Cette étude porte sur le suivi de la population pédiatrique d'un seul ophtalmologiste dans un territoire de santé limité, avec une quasi-exclusivité. En d'autres termes, pratiquement tous les enfants de ce territoire de santé ont été vus par cet ophtalmologiste.
- – Le cycloplégique utilisé est le cyclopentolate, sauf pour les très jeunes enfants.
- – La méthode de réfraction utilisée est la skiascopie.
- – La population comprend des sujets normaux et des sujets strabiques qui ont été étudiés ensemble ou séparément.
- – La durée de l'étude a été la totalité d'un exercice professionnel, c'est-à-dire pendant plus de 30 ans.
Les études statistiques sont des études à la fois longitudinales et transversales, la composante longitudinale étant beaucoup plus forte que la composante transversale. Si tous les sujets étudiés ont bien été inclus dans l'étude dès les premières mesures, le hasard des contrôles et les événements de vie ont fait que chaque groupe étudié ultérieurement est un prélèvement aléatoire dans la population initiale.
Les résultats statistiques sont nombreux et fort riches [5].
Celle-ci va de la naissance à un an [3].
Comme le montre la figure 28-33, il existe une emmétropisation rapide de la sphère méridienne (sphère la plus hypermétrope. Elle correspond à la sphère de base additionnée du cylindre exprimé de manière positive).
« Ce travail a donc permis de confirmer l'existence d'une emmétropisation nette et rapide portant essentiellement sur la régression de l'hypermétropie, le cylindre ayant globalement assez peu d'évolution. Il apparaît tout aussi nettement que cette emmétropisation n'est que partielle laissant pour compte un nombre relativement élevé de fortes amétropies mais aussi de cylindres. Le second point important est que contrairement à certaines affirmations l'écart-type diminue faiblement pendant cette période.
L’essentiel de l’emmétropisation clinique survient entre 3 et 9 mois » [5].
Celle-ci couvre deux phases [3, 5] :
- – l'emmétropisation « secondaire ». Elle correspond à l'évolution entre 1 à 7 ans;
- – l'emmétropisation « tardive ». Elle correspond à l'évolution entre 7 et 20 ans.
« L'ajustement fonctionnel se poursuit lentement jusqu'entre 6 à 7 ans et encore plus discrètement jusqu'entre 15 à 20 ans. La survenue des myopies précoces perturbe de façon notable l'interprétation de cette évolution et conduit nécessairement à plusieurs types de description selon que l'on veut ou non prendre en compte cette dérive myopique. Cette constatation est apparemment partagée par plusieurs auteurs qui, au cours de ces dernières années, ont réalisé des analyses différentielles en fonction des réfractions initiales et des modalités évolutives qui leur sont propres » [16, 33].
« La preuve statistique de l'emmétropisation repose avant tout sur des données partiellement erronées. Le premier obstacle est certainement celui de recrutement biaisé comme le montrent des valeurs très différentes d'une étude à l'autre. L'introduction de populations myopes ou à tendance génétique myopique accentue probablement ces dérives. Mais on constate également que sur des populations homogènes et a priori représentatives comme celle qui a fait l'objet de nos études, il existe des biais de sélection favorisant les réfractions non physiologiques, tant en étude transversale que longitudinale. Il est finalement apparu qu'à partir de l'âge de 3 ans le risque d'erreur lié à ces biais était au minimum du même ordre que la variation que l'on cherche à mesurer. L'élévation sensible de l'écart-type constatée à partir de 6 ans est typiquement liée à la sélection des myopies débutantes » [5].
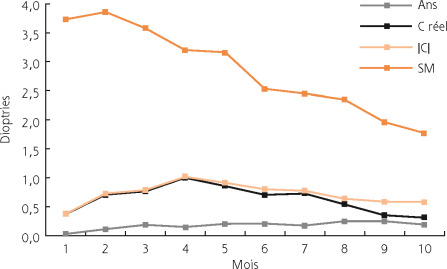
Fig. 28-33 Évolution globale des paramètres de 1 à 10 mois [5].
Le phénomène d'emmétropisation est un phénomène ayant de multiples facettes. Volontairement et pour des raisons de simplicité, nous nous sommes limité aux données consacrées à l'emmétropisation chez l'homme. Malgré ces limitations, nous nous sommes rapidement heurté à la fois à un trop-plein et à un manque de données.
Par ailleurs, ce concept présente deux facettes bien différentes : la tendance vers zéro de la moyenne des amétropies et une éventuelle distribution leptokurtique des réfractions centrée sur une hypermétropie faible. Si une certaine tendance à la leptokurticité semble exister, la tendance vers une moyenne égale à zéro de l’ensemble des réfractions est un phénomène difficile à évaluer du fait de l’irruption du phénomène myopique. Pour les uns, c’est un dérèglement du phénomène d’emmétropisation ; pour d’autres, il fait partie du phénomène d’emmétropisation.
Cependant, un point se dégage avec certitude. À partir d'aujourd'hui, l'anomalie réfractive ne peut plus être étudiée que sur les paramètres optiques; elle doit être étudiée également et surtout sur les paramètres biométriques. Par ailleurs, l'étude du rapport longueur axiale/rayon de courbure (AL/CR) semble une voie intéressante pour dépister les enfants amétropes [15]. C'est sans aucun doute une voie à évaluer.
[1] Attebo K, Ivers RQ, Mitchell P Refractive errors in an older population : the Blue Mountains Eye Study Ophthalmology: ( 1999 ) : 106: 1066-1072
[2] Brown E Net average yearly changes in refraction of atropinized eyes from birth to beyond middle life Arch Ophthalmol: ( 1938 ) : 19: 719-734
[3] Clergeau G. Évolution de la réfraction. In : Péchereau A, Denis D, Speeg-Schatz C. Strabisme. Rapport à la Société Française d’Ophtalmologie, Paris, 2013.
[4] Clergeau G La réfraction de l'enfant: FNRO édition ( 2014 ) 475-
[5] Clergeau G. La réfraction de l’enfant. Péchereau A, Péchereau J (Eds). FNRO édition ; 2014. p. 392. www.larefraction.net/Documents/Ref-Enfant/Ref-Enfant.html.
[6] Curtin BJ The components of refraction and their correlation. The myopias. Basic Science and Clinical Management: Harper & Row ( 1985 ) 17-27
[7] Echard M, Luco C. Biométrie et réfraction. Mémoire de fin d’étude d’Orthoptie, Nantes, p. 21.
[8] Fledelius HC, Stubgaard M Changes in refraction and corneal curvature during growth and adult life : a cross-sectional study Acta Ophthalmol (Copenh): ( 1986 ) : 64: 487-491
[9] Fledelius HC Corneal curvature radius : oculometric considerations with reference to age and refractive change Acta Ophthalmol Suppl: ( 1988 ) : 185: 74-77
[10] Flitcroft DI A model of the contribution of oculomotor and optical factors to emmetropization and myopia Vision Res: ( 1998 ) : 38: 2869-2879
[11] Ganguilhem G. Le normal et le pathologique. Paris : PUF ; 2013.
[12] Grosvenor T, Scott R Role of the axial length/corneal radius ratio in determining the refractive state of the eye Optom Vis Sci: ( 1994 ) : 71: 573-579
[13] Gwiazda J, Marsh-Tootle WL, Hyman L Baseline refractive and ocular component measures of children enrolled in the correction of myopia evaluation trial (COMET) Invest Ophthalmol Vis Sci: ( 2002 ) : 43: 314-321
[14] Hashemi H, Khabazkhoob M, Miraftab M Axial length to corneal radius of curvature ratio and refractive errors J Ophthalmic Vis Res: ( 2013 ) : 8: 220-226
[15] He X, Zou H, Lu L Axial length/corneal radius ratio : association with refractive state and role on myopia detection combined with visual acuity in Chinese schoolchildren PLoS ONE: ( 2015 ) : 10: e0111766-
[16] Jones LA, Mitchell GL, Mutti DO Comparison of ocular component growth curves among refractive error groups in children Invest Ophthalmol Vis Sci: ( 2005 ) : 46: 2317-2327
[17] Kempen JH, Mitchell P, Lee KE The prevalence of refractive errors among adults in the United States, Western Europe, and Australia Arch Ophthalmol: ( 2004 ) : 122: 495-505
[18] Larsen JS The sagittal growth of the eye Acta Ophthalmol (Copenh): ( 1971 ) : 49: 239-262 427-40, 441-53, 873-86
[19] Lawrence MS, Azar DT Myopia and models and mechanisms of refractive error control Ophthalmol Clin North Am: ( 2002 ) : 15: 127-133
[20] Ojaimi E, Rose KA, Morgan IG Distribution of ocular biometric parameters and refraction in a population-based study of Australian children Invest Ophthalmol Vis Sci: ( 2005 ) : 46: 2748-2754
[21] Péchereau A Méthodes instrumentales de mesure de la réfraction La réfraction de l'œil: Elsevier Masson ( 2007 ) 122-125
[22] Rémy C Notions générales d'optique et optique de l'œil La réfraction de l'œil: Elsevier Masson ( 2007 ) 3-19
[23] Rosenbaum R Brit J Industr Med: ( 1957 ) : 14: - cité par [25]
[24] Slataper F Age norms of refraction and vision Arch Ophthalmol: ( 1950 ) : 43: 466-481
[25] Sorsby A, Sheridan M, Leary GA, Benjamin B Vision, visual acuity, and ocular refraction of young men : findings in a sample of 1,033 subjects Br Med J: ( 1960 ) : 1: 1394-1398
[26] Steiger A. Die Entsehung der sphärischen Refractionen des Auges. Berlin : S. Karger ; 1913.
[27] Straub M Over de Aetiologie der Brekingsafwijkingen van het Oog en den Oorsprung der Emmetropie Nederlandsch Tijdschrift voor Geneskunde: ( 1909 ) : 7-9: 445-460 533-56, 639-64
[28] Touzeau O, Allouch C, Borderie V Corrélation entre la réfraction et la biométrie oculaire J Fr Ophtalmol: ( 2003 ) : 26: 355-363
[29] Touzeau O Cahiers d'Ophtalmologie: ( 2015 ) : 189: 14-19
[30] Wong TY, Foster PJ, Johnson GJ, Seah SK Refractive errors, axial ocular dimensions, and age-related cataracts : the Tanjong Pagar survey Invest Ophthalmol Vis Sci: ( 2003 ) : 44: 1479-1485
[31] Zadnik K, Manny RE, Yu JA Ocular component data in schoolchildren as a function of age and gender Optom Vis Sci: ( 2003 ) : 80: 226-236
[32] Zadnik K, Mutti DO, Friedman NE, Adams AJ Initial cross-sectional results from the Orinda Longitudinal Study of Myopia Optom Vis Sci: ( 1993 ) : 70: 750-758
[33] Zadnik K The Glenn A. Fry Award Lecture (1995). Myopia development in childhood Optom Vis Sci: ( 1997 ) : 74: 603-608