Tumeurs de la surface oculaire
Les tumeurs de la surface oculaire sont de nature extrêmement variée. En effet, l’ensemble des éléments constituant l’épithélium et le stroma conjonctival peut être à l’origine d’un processus tumoral. Au niveau de la cornée, les tumeurs seront essentiellement d’origine épithéliale, les tumeurs stromales étant beaucoup plus rares. La caroncule, structure particulière constituée à la fois d’éléments cutanés, muqueux et glandulaires, pourra présenter des tumeurs d’origine épithéliale ou stromale mais aussi d’origine cutanée ou glandulaire.
Au niveau de la surface oculaire, les tumeurs bénignes sont beaucoup plus fréquentes que les tumeurs malignes, plus rares, essentiellement représentées par les néoplasies de type épidermoïde, le lymphome et le mélanome.
Ce chapitre regroupe l’ensemble des tumeurs touchant la surface oculaire dont toutes les composantes peuvent être impliquées.
Ainsi, les tumeurs peuvent être à point de départ : épithélial le plus souvent, stromal plus rarement, mais aussi mélanocytaire, vasculaire ou lymphoïde. Si la nature bénigne reste la plus fréquente, certaines d’entre elles possèdent un haut grade de malignité et doivent ainsi pouvoir bénéficier d’un diagnostic rapide et d’une prise en charge adaptée. Parmi ces tumeurs malignes, les lésions næviques arrivent en tête et représentent plus d’un quart de l’ensemble des tumeurs conjonctivales dans la série publiée par Shields [1].
La prise en charge thérapeutique des tumeurs conjonctivales est différente selon la nature histologique de la tumeur. Il est donc utile d’avoir identifié la lésion avant tout geste chirurgical pour adapter le type d’exérèse (exérèse complète en bloc de la tumeur ou simple biopsie), l’anesthésie (locale ou générale) et le degré d’urgence de la chirurgie à la nature de la tumeur. En cas de suspicion de lymphome, on ne pratiquera que des prélèvements biopsiques sans chercher à réaliser une exérèse complète. En cas de suspicion de mélanome ou de carcinome épidermoïde, il faudra par contre prévoir l’exérèse complète de la lésion, rapidement et sous anesthésie générale. Une « no touch » technique est recommandée : manipulation minimum de la tumeur ; utilisation d’alcool absolu pour décoller l’épithélium cornéen ; cautérisation des vaisseaux nourriciers ; exérèse de la tumeur en bloc avec marges latérales de quelques millimètres ; changements d’instruments pour la reconstruction [2, 3]. La pièce d’exérèse devra si possible être orientée et étalée sur du papier ou sur une éponge afin d’éviter qu’elle ne s’enroule sur elle-même. En cas de mélanome ou de carcinome épidermoïde, il sera aussi indispensable de noter toutes les informations possibles sur la localisation et les dimensions tumorales juste avant la chirurgie pour faciliter la réalisation d’une éventuelle irradiation complémentaire (réalisation de photos en lampe à fente et d’un schéma de la tumeur précisant les horaires au limbe et les diamètres tumoraux en millimètre). Pour toutes les tumeurs malignes, la prise en charge thérapeutique sera discutée en réunion de concertation pluridisciplinaire.
Les tableaux 11-1 et 11-2 résument les principaux diagnostics à évoquer devant un aspect de tumeur achrome (voir tableau 11-1) ou pigmentée (voir tableau 11-2), en fonction du terrain, de l’évolution et des caractéristiques cliniques de la lésion.
L. Desjardins
Mis à part le papillome, les tumeurs épithéliales (qu’elles soient bénignes ou malignes) apparaissent essentiellement sur des patients d’âge adulte. Le développement pendant l’enfance de tumeurs épithéliales malignes est tout à fait exceptionnel et se voit essentiellement chez des enfants présentant un xeroderma pigmentosum (XP). Le xeroderma pigmentosum (fig. 11-1) correspond en fait à un groupe d’affections héréditaires de transmission autosomique récessive entraînant un défaut de la réparation de l’acide désoxyribonucléique (ADN), en particulier au niveau des cellules cutanées exposées aux effets mutagènes des rayons ultraviolets [4, 5]. Cette maladie grave et invalidante, mettant en jeu le pronostic esthétique, visuel et vital, est très rare en Europe et aux États-Unis (prévalence 1/1 000 000), plus fréquente au Maghreb (1/10 000 en Tunisie) et au Moyen-Orient où le pourcentage de mariages consanguins est important [6]. Le tableau clinique est dominé par des manifestations cutanées et oculaires d’hypersensibilité au soleil. L’apparition inéluctable de cancers cutanéomuqueux, avec un risque 1 000 à 4 800 fois plus élevé que dans la population générale [7], en fait toute la gravité, raccourcissant notablement l’espérance de vie des patients [8, 9]. La maladie débute souvent dans l’enfance avant 10 ans. Sur le plan ophtalmologique, on retrouve une conjonctivite et une photophobie (symptôme le plus fréquent retrouvé dans 66 % des cas [10]) avec un syndrome sec et parfois une insuffisance limbique. Une mélanose conjonctivale peut aussi être présente. Les cicatrices cornéennes et la néovasculariastion aboutissent souvent à une baisse d’acuité visuelle [11]. Les néoplasies de la surface oculaire rencontrées vont de la dysplasie modérée à sévère au carcinome in situ et au carcinome invasif. Elles sont retrouvées selon les séries entre 10 % des cas [11] et plus de 70 % des cas [12]. Sur le plan génétique, il a été démontré que la variabilité clinique du XP répondait à sept mutations germinales différentes (A, B, C, D, E, F et G), le XP variant représentant une forme particulière. En pratique, il n’existe pas de correspondance absolue entre les sept formes génétiques de la maladie et le tableau clinique réalisé. La prise en charge thérapeutique pose souvent des problèmes thérapeutiques complexes ; elle repose essentiellement sur des mesures de prévention avec en particulier une surveillance ophtalmologique rapprochée dès l’enfance afin de traiter au mieux le syndrome sec et de diagnostiquer et traiter précocement les lésions dysplasiques de la surface oculaire. Le but étant d’éviter au maximum l’apparition de carcinomes invasifs et l’utilisation de la radiothérapie. Celle-ci est cependant parfois inévitable pour permettre la conservation oculaire. La qualité de vie des patients est médiocre et le pronostic est globalement sombre (les deux tiers des malades meurent avant d’atteindre l’âge adulte).

Tableau 11-1 Principales tumeurs achromes de la surface oculaire et de la conjonctive palpébrale.

Tableau 11-2 Principales tumeurs pigmentées de la surface oculaire et de la conjonctive palpébrale.

Fig. 11-1 Xeroderma pigmentosum avec multiples lésions tumorales carcinomateuses au niveau du visage, des paupières et de la surface oculaire.
Le papillome conjonctival est une tumeur bénigne d’origine virale qui atteint plus fréquemment les enfants et les adultes jeunes. L’incidence la plus élevée se situe entre 20 et 39 ans. C’est une tumeur rare représentant environ 0,5 à 2,5 % de l’ensemble des tumeurs conjonctivales [1]. La relation causale avec la présence d’un papillomavirus humain (human papilloma virus ou HPV) a été documentée dans 5 à 45 % des cas. Il s’agit le plus souvent de virus HPV à faible risque de carcinogenèse (types 6 et 11). Plus rarement, en particulier chez l’adulte, il s’agit de virus HPV à haut risque (types 16, 18 et 33). L’identification de l’HPV en cause peut se faire par polymerase chain reaction (PCR) après extraction de l’ADN sur du tissu congelé [13, 14].
Les caractéristiques cliniques diffèrent chez l’enfant et chez l’adulte.
Le papillome conjonctival de l’enfant peut être solitaire ou multiple (les formes multicentriques étant plus fréquentes chez l’enfant et l’adolescent que chez l’adulte). Il se présente sous la forme d’une excroissance rosée d’aspect framboisé souvent pédiculée (fig. 11-2). Cet aspect framboisé est dû à la présence de très nombreux capillaires au niveau du stroma sous l’épithélium. Très rarement, plusieurs lésions peuvent confluer et produire une papillomatose massive. Le siège de prédilection est le cul-de-sac ou la conjonctive bulbaire ; la lésion déborde rarement sur la cornée. Les papillomes de l’enfant sont associés aux sous-types 6 et 11 du papillomavirus humain. Sur le plan histologique, on retrouve des papilles fibrovasculaires recouvertes d’un épithélium malpighien hyperplasique. Il y a en général peu ou pas de kératinisation mais le nombre de cellules caliciformes est parfois diminué. On peut retrouver des signes d’inflammation chronique avec présence de polynucléaires neutrophiles. L’évolution est bénigne. En cas de papillome irritant, inesthétique, ou en cas de doute diagnostique, une chirurgie d’exérèse peut être proposée. Dans ce cas, l’exérèse se doit d’être complète pour éviter les récidives, et il est conseillé lors de l’exérèse d’éviter de toucher le papillome lui-même afin de ne pas libérer de particule virale dans les tissus avoisinants. Des traitements adjuvants locaux ou par voie orale sont possibles en cas de papillome récidivant ou invasif : citons la cryothérapie, le laser, l’immunothérapie par le dinitrochlorobenzène, les collyres à l’interféron [15] ou à la mitomycine [16]. Une réponse spectaculaire à la cimétidine par voie orale a aussi été décrite [17, 18].
Chez l’adulte, le papillome conjonctival est plus souvent sessile et plus souvent localisé au niveau de la caroncule (le papillome représentant selon les études 13 à 31 % des lésions retrouvées au niveau de la caroncule [19]). Il peut aussi se développer au niveau de la conjonctive bulbaire et recouvrir partiellement la cornée. Il est généralement unique, de coloration un peu plus rose pâle que chez l’enfant et il peut se pigmenter chez les patients mélanodermes. Cliniquement, le papillome de l’adulte peut être difficile à différencier d’une néoplasie épidermoïde (carcinome in situ ou invasif) [20] ou d’un mélanome achrome, sachant qu’au niveau du limbe chez l’adulte, les néoplasies épidermoïdes de la surface oculaire sont presque 10 fois plus fréquentes que les papillomes [21]. Les papillomes sessiles sont parfois associés aux types 16 et 18 du virus HPV mais aussi souvent aux types 6 et 11 [22]. La présence d’une inflammation et d’une leucoplasie doit faire suspecter la possibilité d’une transformation maligne. Le plus souvent, une biopsie exérèse est nécessaire pour avoir un diagnostic histologique précis. Lors de l’exérèse chirurgicale, la partie cornéenne, s’il y en a une, se clive très facilement, la base de la lésion étant située au niveau du limbe. Histologiquement, le papillome sessile a une base large avec un épithélium épaissi mais pas d’anomalies cytonucléaires. Une hyperkératose modérée est souvent présente. Une cryothérapie des berges est parfois réalisée. Une récidive est possible. L’évolution vers une tumeur maligne bien que rare a été décrite [23].

Fig. 11-2 Papillome conjonctival du cul-de-sac inférieur.
Le kératoacanthome est une lésion bénigne rare. Sur 195 lésions caronculaires, Kaeser [24] retrouve seulement un kératoacanthome. Cette tumeur isolée intéresse souvent le bord libre de la paupière mais peut occasionnellement se développer au niveau de la caroncule ou de la conjonctive bulbaire. Elle se caractérise par une croissance rapide en quelques semaines. La tuméfaction rapidement très en relief présente une invagination centrale contenant des débris de kératine. Bien que la croissance rapide et l’aspect clinique puissent faire suspecter une lésion carcinomateuse, ces lésions régressent spontanément en quelques mois avec une cicatrice minime. Histologiquement, on retrouve une dépression centrale remplie de débris de kératine. La paroi de la lésion est délimitée par un épithélium stratifié épaissi et kératinisé. Il est parfois difficile de distinguer cette lésion d’une forme superficielle de carcinome épidermoïde bien différencié, c’est pourquoi on dit qu’il s’agit d’une forme d’hyperplasie pseudo-épithéliomateuse. En général, il n’y a pas d’atteinte du chorion en profondeur, comme c’est le cas dans les carcinomes épidermoïdes, et pas de véritables atypies cellulaires, en dehors de dysplasies minimes associées. Il est cependant souvent nécessaire de réaliser une exérèse chirurgicale de ces lésions en raison de la suspicion de tumeur maligne [25].
L’hyperplasie pseudo-épithéliomateuse est une réaction non spécifique à un changement des tissus avoisinants et en particulier à un processus inflammatoire réalisant une masse pseudo-tumorale. L’inflammation sous-jacente peut en effet induire une hyperplasie de l’épithélium qui peut cliniquement être confondue avec un carcinome. Histologiquement, l’épithélium conjonctival est épaissi et acanthosique avec présence de parakératose et parfois d’une kératinisation. Les cellules épithéliales ne présentent pas de dyskératose majeure ou de pléomorphisme cellulaire, ce qui différencie cette lésion d’un carcinome.
La kératose folliculaire inversée est une lésion rare, bien circonscrite ressemblant à un papillome qui intéresse plus souvent la peau des paupières que la conjonctive. Comme dans le kératoacanthome, la croissance rapide peut faire suspecter une tumeur maligne. Sur le plan histologique, cette lésion est considérée comme une variante de l’hyperplasie pseudo-épithéliomateuse. On retrouve une acanthose, une parakératose et des couches concentriques de cellules pavimenteuses.
La dyskératose héréditaire bénigne est une maladie autosomale dominante caractérisée par une dyskératose de la conjonctive, de la cornée et de l’épithélium buccal. La lésion conjonctivale bilatérale est située en temporal ou en nasal dans l’aire de la fente palpébrale. L’aspect est de forme triangulaire ou irrégulière avec une surface surélevée et une vascularisation importante. Elle apparaît en général lors de la première décade. Elle peut rester asymptomatique ou être responsable d’une hyperhémie sévère et d’une sensation de corps étranger. Histologiquement, il existe un épaississement irrégulier de l’épithélium avec acanthose et dyskératose mais la membrane basale est intacte. L’aspect le plus caractéristique est la kératinisation et l’existence d’une inflammation chronique non granulomateuse en surface. Le traitement peut se limiter à des collyres lubrifiants et corticoïdes, mais les lésions les plus volumineuses doivent parfois être enlevées chirurgicalement. Une exérèse chirurgicale associée à une greffe de cellules limbiques est proposée par Cai [26].
Les plaques de kératose sont des lésions dégénératives. Le risque de transformation maligne en carcinome épidermoïde invasif est très faible. Cliniquement, les plaques de kératose sont plus fréquentes dans l’aire de la fente palpébrale, près du limbe, dans la zone conjonctivale exposée au vent et au soleil. L’incidence est plus élevée dans les pays chauds et elles peuvent parfois apparaître à la surface d’un ptérygion. L’aspect clinique est celui d’une lésion plane de coloration blanchâtre avec une surface rugueuse qui apparaît progressivement et se développe lentement. Il existe parfois une dilatation des vaisseaux conjonctivaux à la périphérie de ces lésions. Le retentissement fonctionnel est en général modéré. La plaque de Bitot dans les hypovitaminoses A est un exemple de plaque de kératose focale circonscrite [27].
Sur le plan histologique, les plaques de kératose sont des lésions acanthosiques avec kératinisation de l’épithélium de surface et parakératose. Les atypies sont minimes ou inexistantes contrairement à ce qui est observé dans les carcinomes in situ. Une forme particulière, la kératose séborrhéique, a été rarement décrite au niveau de la conjonctive. Elle comporte alors des pseudo-kystes au sein de l’épithélium épaissi [28]. La prise en charge thérapeutique consiste généralement en une exérèse simple sous anesthésie locale avec examen histologique afin de redonner un aspect satisfaisant à la conjonctive bulbaire limbique et d’éliminer toute possibilité de lésion carcinomateuse. Chez des patients âgés en mauvais état général, en l’absence de progression notable de la lésion, une simple surveillance est parfois justifiée.
La kératose actinique est une plaque de kératose induite par l’exposition aux rayons ultraviolets. Cliniquement, une leucoplasie est souvent présente comme dans les plaques de kératose avec un aspect en « sucre mouillé » (fig. 11-3). Sur le plan histologique, il existe un pléomorphisme cellulaire, une dyskératose et des mitoses. On note aussi une dégénérescence élastinique du collagène caractéristique des dommages causés par les rayons ultraviolets [29]. Il n’y a pas de franchissement de la basale mais la kératose actinique peut évoluer vers un carcinome invasif.
Récemment en 2013, des granulomes actiniques ont été décrits chez des femmes jeunes [30] ou moins jeunes [31] exposées au soleil. Ils seraient secondaires à la destruction des fibres de collagène et s’accompagnent de la présence de cellules géantes mais sans hyperkératose de surface. Le traitement de toutes ces lésions est chirurgical par exérèse simple sous anesthésie locale avec examen histologique.

Fig. 11-3 Kératose actinique du chorion sous-jacent.
Le terme de néoplasies épidermoïdes de la surface oculaire regroupe l’ensemble des lésions épidermoïdes précancéreuses et cancéreuses de la conjonctive et de la cornée. Les dysplasies et les carcinomes in situ sont des néoplasies épidermoïdes uniquement intra-épithéliales (la prolifération tumorale respecte la membrane basale et n’envahit pas le chorion sous-jacent), il s’agit donc de lésions précancéreuses. Ce sont des lésions rares atteignant les adultes de toutes les races. À partir de 20 ans, elles représenteraient environ 10 % des lésions conjonctivales [32]. Elles sont plus fréquentes à partir de 55-60 ans et dans les pays proches de l’équateur et en cas d’importante exposition aux rayons ultraviolets. L’incidence est de 0,03 pour 100 000 aux États-Unis, 0,13 pour 100 000 en Ouganda et 1,9 pour 100 000 en Australie. L’exposition aux rayons ultraviolets est un facteur de risque majeur et le risque augmente avec le temps d’exposition, la vie en extérieur et chez les sujets à peau claire et à iris clair [33]. Des mutations liées aux rayons ultraviolets au niveau de gène suppresseur de tumeurs comme le P53 ont été retrouvées [34]. La protéine SIRT-1, impliquée dans la réparation de l’ADN, est surexprimée dans ces lésions [35]. La présence de virus HPV a été détectée chez des patients porteurs de néoplasies épidermoïdes de la surface oculaire mais aussi chez des patients indemnes [36]. Son rôle n’est pas clairement déterminé, mais il agirait plus comme cofacteur que comme cause véritable.
Par contre, l’existence d’une infection par le virus de l’immunodéficience humaine (VIH) est très fortement associée à une augmentation des néoplasies épidermoïdes de surface qui atteignent alors des sujets plus jeunes et revêtent une gravité particulière avec une évolution beaucoup plus rapide. Dans notre expérience, l’utilisation de collyre à la ciclosporine favorise également le développement de ces lésions.
Sur le plan clinique, les néoplasies épidermoïdes intra-épithéliales (dysplasies et carcinomes in situ) apparaissent comme des lésions planes ou légèrement surélevées, sessiles, développées le plus souvent dans l’aire d’ouverture de la fente palpébrale, plus rarement dans les culs-de-sac ou sur la conjonctive palpébrale. Elles sont retrouvées dans 1,7 % des cas à la surface d’un ptérygion [37]. Toute lésion conjonctivale enlevée chirurgicalement doit donc être adressée au laboratoire pour examen anatomo-pathologique. Ces lésions sont en général achromes mais elles peuvent présenter une pigmentation hétérogène chez les patients mélanodermes. L’aspect est parfois papillomateux, parfois leucoplasique ou les deux associés (fig. 11-4 et 5a). L’envahissement cornéen lorsqu’il est présent a souvent un aspect gélatineux (fig. 11-5b). Une inflammation peut être associée et orienter à tort vers une conjonctivite. Un aspect papillomateux ou nodulaire serait en faveur d’une lésion de haut grade [38]. L’existence d’une leucoplasie, d’une épaisseur, l’absence d’envahissement cornéen et une importante vascularisation seraient plutôt en faveur d’un carcinome invasif mais il n’y a pas de critères cliniques clairs pour différencier les lésions purement intra-épithéliales (dysplasie et carcinome in situ) des lésions ayant envahi le chorion (carcinome invasif). Le diagnostic de certitude est histologique : on retrouve un épithélium hyperplasique avec perte de la polarité cellulaire normale, hyperchromatisme nucléaire et pléomorphisme ; des figures mitotiques peuvent être présentes. Il y a souvent une kératinisation de surface, en rapport avec l’aspect leucoplasique visible cliniquement. Une dyskératose (présence de cellules produisant de la kératine au niveau des couches profondes) peut aussi se voir ainsi qu’une réaction inflammatoire au niveau du chorion. Le point le plus important de l’histologie est le caractère strictement intra-épithélial de la prolifération qui ne dépasse pas la membrane basale en profondeur. Cette néoplasie ou dysplasie peut être classée en débutante, modérée (fig. 11-6a) ou sévère, en fonction du degré d’atypies cellulaires. Lorsque les atypies sont très nombreuses et sévères, et que la lésion s’étend sur toute l’épaisseur de l’épithélium, le terme de carcinome in situ doit être utilisé (fig. 11-6b).
Le traitement habituel consiste en une exérèse chirurgicale complète de la lésion [39]. Il faut éviter en enlevant la partie cornéenne d’altérer la membrane de Bowman. L’utilisation d’alcool absolu permet en général un clivage facile. Dans les cas où la lésion cornéenne est trop étendue, nous préconisons d’enlever seulement la partie conjonctivale de la lésion et de traiter le reliquat cornéen ensuite avec des collyres chimiothérapiques. Une exérèse incomplète avec des berges positives est un facteur majeur de récidive locale [40, 41]. La cryothérapie des berges a été très largement utilisée. Elle réduirait le taux de récidive locale [42]. Actuellement, on privilégie surtout l’utilisation des chimiothérapies topiques en collyres (mitomycine C, interféron, 5-fluorouracile), extrêmement efficaces dans cette pathologie [43]. Ces collyres sont utilisés en cas d’exérèse incomplète et/ou en cas de récidive locale. Pour les effets secondaires et complications se référer au chapitre 4. Soulignons qu’il est important de bien différencier les récidives de carcinome in situ des altérations secondaires aux insuffisances limbiques, parfois provoquées par les collyres antimitotiques, afin d’utiliser ces derniers à bon escient.
La mitomycine est le collyre le plus couramment employé dans le traitement adjuvant des carcinomes in situ [43]. La technique a été décrite en 1997 par Frucht-Pery [44] et de nombreuses publications attestent de son efficacité [45-51]. Certains auteurs l’utilisent aussi comme traitement de première intention [52]. À l’Institut Curie, les protocoles varient selon les indications : en traitement complémentaire après exérèse chirurgicale complète d’un carcinome in situ, nous utilisons le collyre à la mitomycine dosé à 0,02 % et nous réalisons deux cures de 15 jours (une goutte 4 fois/jour), les deux cures étant espacées de 15 jours d’arrêt (fig. 11-7). En cas de rechute, ou d’exérèse incomplète, nous préconisons alors l’utilisation de collyre à la mitomycine dosé à 0,04 % en cures de 8 jours espacées de 8 jours, mais la toxicité cornéenne est alors plus importante et justifie une surveillance ophtalmologique pendant le traitement.
Le 5-fluorouracile peut être utilisé en collyre à 1 %. La première utilisation dans les carcinomes in situ a été décrite par De Keiser [53]. Les travaux de Midena et Parrozzani [54, 55] ont confirmé son efficacité et son innocuité sur des patients ayant un carcinome invasif récidivant ou partiellement excisé. La posologie utilisée était d’une goutte 1 fois/mois pendant 1 mois suivie de 3 mois sans traitement en répétant les cycles jusqu’à rémission complète. Un taux de rechute locale d’environ 15 % était noté. À l’Institut Curie, nous n’utilisons pas les chimiothérapies topiques pour les carcinomes invasifs, nous préférons utiliser la radiothérapie (voir plus loin Carcinome épidermoïde invasif).
L’interféron alpha est employé dans le traitement du carcinome in situ depuis 1994 [56]. Karp a rapporté un traitement efficace par l’interféron alpha utilisé en continu pendant plusieurs mois chez cinq patients [57]. Une dose d’un million d’unités par millilitre est généralement prescrite 4 fois/jour. Besley [58] recommande son utilisation dans les carcinomes non invasifs (in situ). La résolution des carcinomes in situ nécessite en général environ 3 mois de traitement. L’efficacité de l’interféron alpha associé parfois à de l’acide rétinoïque [59] dans les carcinomes in situ est rapportée par plusieurs auteurs : Huerva [60], Schechter [61], Shah [62]. Plus récemment, Shields a rapporté l’utilisation de l’interféron alpha dans 82 cas de néoplasies de la surface oculaire incluant 52 cas de forme invasive [63].
Au total, le traitement des carcinomes in situ par les collyres antimitotiques a l’avantage de traiter l’ensemble de la surface oculaire, y compris les cellules atypiques non détectables cliniquement, et d’éviter les effets secondaires de la chirurgie. Bien qu’il n’y ait pas d’étude randomisée comparant les différents traitements, la revue des études publiées dans la littérature montre une efficacité similaire entre les trois agents. La résolution des lésions est plus rapide avec la mitomycine, mais c’est aussi la drogue qui a le plus d’effets secondaires. L’interféron alpha a moins d’effets secondaires mais est aussi beaucoup plus cher. Étant donné qu’une récidive de ces lésions peut apparaître tardivement, parfois plusieurs années après le traitement, le suivi de ces patients est essentiel pour bien évaluer l’efficacité des traitements et leurs effets secondaires.

Fig. 11-4 Aspect clinique papillomateux d’une dysplasie conjonctivale avec atypies cellulaires modérées en histologie.

Fig. 11-5 Aspect clinique de carcinome in situ.
Aspects papillomateux (a) et gélatineux d’une récidive limbique envahissant la périphérie cornéenne (b).

Fig. 11-6 Aspects histologiques des néoplasies épidermoïdes de la surface oculaire.
a. Dysplasie conjonctivale avec atypies modérées : présence d’atypies cellulaires plus ou moins marquées n’intéressant que les couches profondes de l’épithélium, les couches les plus superficielles étant normales.
b. Carcinome épidermoïde in situ : les atypies sont plus sévères et intéressent toute l’épaisseur de l’épithélium, mais elles respectent la membrane basale.
c. Carcinome épidermoïde invasif : les anomalies sont similaires au carcinome épidermoïde in situ mais avec franchissement de la membrane basale et envahissement du chorion sous-jacent.

Fig. 11-7 Carcinome in situ.
Aspect avant (a) et après traitement par exérèse chirurgicale et collyre mitomycine (b).
Le carcinome épidermoïde invasif fait partie des néoplasies épidermoïdes de la surface oculaire. Il se différencie cependant des néoplasies épidermoïdes précancéreuses précédemment décrites (dysplasie et carcinome in situ) par la présence d’un envahissement tumoral en profondeur au niveau du chorion (voir fig. 11-6c). Le carcinome épidermoïde est donc une tumeur cancéreuse à potentiel métastatique. Il dérive souvent d’une lésion précancéreuse comme la kératose actinique ou le carcinome in situ. L’exposition aux rayons ultraviolets a été reconnue comme facteur de risque [64], de même que le rôle possible des infections à virus HPV [65, 66]. Pour Chauhan [67] l’association à un virus HPV-16 serait retrouvée dans des tumeurs de meilleur pronostic. La maladie atteint plus fréquemment les hommes après 60 ans [68]. En cas d’infection par le VIH, elle peut toucher aussi les sujets plus jeunes [69-71], elle est alors beaucoup plus agressive.
Cliniquement, le carcinome épidermoïde invasif se présente sous la forme d’une lésion en relief multilobulée, souvent située près du limbe et débordant sur la cornée (fig. 11-8a et 11-9a). Les vaisseaux nourriciers sont dilatés. Fréquemment, la masse est gélatineuse ou d’aspect papillomateux avec des degrés variables de leucoplasie et ne peut être différenciée cliniquement d’un carcinome in situ. L’extension de la lésion sous forme d’une invasion épithéliale en nappes se propageant au niveau de la surface oculaire est possible mais moins fréquente que dans les carcinomes sébacés [72]. Les formes ulcérantes sont peu fréquentes [73]. La plupart du temps, la sclère et la membrane de Bowman représentent une barrière protectrice empêchant l’extension intra-oculaire. De ce fait, l’infiltration sclérale et cornéenne ainsi que l’envahissement intra-oculaire sont des complications rares du carcinome épidermoïde invasif. Elles surviendraient surtout par infiltration de la sclère au niveau du trajet d’une vortiqueuse ou au niveau d’une brèche réalisée lors d’une chirurgie antérieure [74, 75]. Ces aspects d’envahissement intra-oculaire sont plus fréquents dans un contexte d’immunodéficience, les tumeurs étant alors plus agressives.
En l’absence de traitement, l’évolution du carcinome épidermoïde se fait localement vers l’orbite, et la dissémination par voie lymphatique se produit au niveau des adénopathies locorégionales. La classification TNM (tumor-nodes-metastases) tient compte du diamètre et de l’épaisseur de la tumeur et de son extension locorégionale [76, 77]. L’envahissement ganglionnaire éventuel doit de toute façon être précisé par positon emission tomography scan (PET scan) ou imagerie par résonance magnétique (IRM) du massif facial.
La caractéristique histologique majeure des carcinomes épidermoïdes invasifs de la conjonctive est le franchissement de la membrane basale et le développement dans le tissu conjonctif sous-épithélial (voir fig. 11-6c). La tumeur est composée essentiellement de cellules épidermoïdes plus ou moins différenciées. On retrouve le pléomorphisme cellulaire, les atypies cytonucléaires et les mitoses, la dyskératose et la kératinisation en surface.
Il existe des formes histologiques plus rares et plus agressives. Le carcinome à cellules fusiformes est une forme histologique rare de carcinome épidermoïde. Les cellules fusiformes sont parfois difficiles à différencier des fibroblastes, d’où l’intérêt d’une immuno-histochimie positive pour la cytokératine affirmant l’origine épithéliale des cellules. Le carcinome épidermoïde adénoïde présente une architecture pseudo-glandulaire avec présence d’acide hyaluronique extracellulaire sans mucine intracellulaire. Son évolution est particulièrement agressive avec fréquentes métastases ganglionnaires et viscérales [78]. Le carcinome muco-épidermoïde est une forme rare très agressive et de mauvais pronostic. La présentation clinique est voisine de celle des carcinomes épidermoïdes, avec une croissance rapide et un caractère invasif marqué [79]. Ces tumeurs sont probablement sous-diagnostiquées car le diagnostic histologique nécessite des colorations spécifiques. Dans une étude portant sur 273 tumeurs conjonctivales, Alves [80] retrouve 42 néoplasies épidermoïdes dont 15 carcinomes in situ, 17 carcinomes épidermoïdes invasifs et seulement un carcinome muco-épidermoïde. Sur le plan histologique, le carcinome muco-épidermoïde contient des cellules épidermoïdes et des cellules muco-sécrétantes [81]. Une sécrétion de mucine peut être mise en évidence. Pour Jastrzebsi [82], les marquages les plus utiles sont la mucicarmine et le bleu alcyan avec une sensibilité de 88 % et une spécificité de 100 %. Les cytokératines 7 et 20 auraient une sensibilité beaucoup moins bonne.
Le traitement chirurgical du carcinome épidermoïde invasif repose sur une exérèse chirurgicale la plus complète possible sous anesthésie générale de préférence. Comme pour toutes les tumeurs conjonctivales, on utilisera une « no touch » technique ; un schéma et des mesures tumorales peropératoires seront réalisés. Shields [83] a rapporté l’utilisation de collyre à la mitomycine pour réduire la tumeur avant l’exérèse chirurgicale. Ceci peut être particulièrement utile pour les tumeurs très étendues ou recouvrant totalement la cornée. La possibilité de réaliser une biopsie du ganglion sentinelle a été évoquée par Maalouf, Savar et Pfeiffer [84-86], mais elle serait surtout utile pour les tumeurs de grande taille ou localement invasives (tumeurs à haut risque métastatique ganglionnaire). Pour les tumeurs adhérentes à la sclère, certains réalisent une dissection lamellaire de la sclère. Néanmoins la marge de sécurité en profondeur risque toujours d’être insuffisante et il nous paraît préférable de faire une exérèse simple suivie de radiothérapie du lit d’exérèse, le risque de récidive étant de 40 % environ en l’absence de traitement complémentaire.
La radiothérapie adjuvante peut se faire par irradiation externe, curiethérapie ou protonthérapie (fig. 11-8b et 11-9b). De nombreux auteurs ont rapporté l’efficacité de la radiothérapie pour réduire au maximum le risque de récidive locale [87-92]. La méthode d’irradiation sera décidée avec le radiothérapeute lors d’une réunion de concertation pluridisciplinaire. La curiethérapie peut être utilisée pour des petites tumeurs ou en cas de contre-indication de la protonthérapie (malade impotent ou psychiatrique). La protonthérapie permet une irradiation qui dessine les contours exacts de la tumeur et donne des résultats très satisfaisants. Une dose de 60 grays équivalent cobalt est délivrée en huit fractions sur 2 semaines. Le taux de rechute locale est de 2 % et la complication la plus fréquente est la cataracte. Il n’y a pas de contre-indications à opérer cette cataracte s’il existe une rémission complète de la tumeur. Des télangiectasies conjonctivales peuvent se développer dans les années qui suivent l’irradiation au niveau du territoire irradié. En dehors de leur aspect parfois un peu inesthétique, elles n’ont pas de conséquences. Si on veut faire bénéficier le patient de ce traitement, il faut absolument éviter une dissection lamellaire de la sclère lors de la chirurgie. Par ailleurs, s’il existe une lésion étendue de carcinome in situ associée au carcinome invasif, elle sera au mieux traitée par des collyres antimitotiques. Seule la zone de carcinome invasif sera irradiée afin de limiter le champ d’irradiation et les complications oculaires. Les tumeurs envahissant le cul-de-sac relèvent plutôt d’une technique d’irradiation externe conventionnelle. La radiothérapie prophylactique des aires ganglionnaires peut aussi se discuter en fonction de l’étendue de la lésion initiale. En cas de carcinomes muco-épidermoïdes, le traitement chirurgical doit consister en une exérèse large [93] car ils sont plus radio-résistants que les carcinomes épidermoïdes. L’évolution est souvent fatale en cas d’envahissement orbitaire.
Pour tous les carcinomes épidermoïdes invasifs, un suivi rapproché sera de toute façon indispensable localement et au niveau des aires ganglionnaires. La dissémination métastatique viscérale est peu fréquente sauf en cas d’atteinte évoluée avec envahissement orbitaire, de syndrome d’immunodéficience acquise (sida) ou de forme histologique particulièrement agressive. Néanmoins, il est impossible de donner des chiffres précis étant donné la rareté de la tumeur et un suivi souvent insuffisant dans les études publiées. Dans notre expérience, seuls les patients positifs au VIH ont développé des métastases viscérales. Certains patients ayant des tumeurs volumineuses ou envahissant l’orbite ont développé des métastases ganglionnaires mais ont guéri avec un traitement chirurgical et radiothérapique. Pour Shields, le risque de métastase ganglionnaire ne serait que de 2 % [68].

Fig. 11-8 Carcinome épidermoïde invasif.
Aspect avant (a) et après traitement par chirurgie et irradiation complémentaire par disque d’iode 125 (b).

Fig. 11-9 Carcinome épidermoïde invasif.
Aspect avant (a) et après traitement par chirurgie et irradiation complémentaire par protons (b).
Les carcinomes basocellulaires primitifs de la conjonctive sont extrêmement rares et se développent plus souvent au niveau de la caroncule [94, 95]. Par contre, l’envahissement de la conjonctive peut survenir lorsqu’il existe un carcinome basocellulaire des paupières surtout en cas de récidive ou dans le syndrome de Gorlin-Goltz. Cette maladie, ou syndrome du nævus basocellulaire, qui se transmet selon un mode autosomal dominant, entraîne un risque accru de carcinome basocellulaire des paupières qui apparaissent à un âge précoce (30 ans). Pour Taylor [96], 61 % des patients atteints de ce syndrome développeraient des carcinomes basocellulaires péri-oculaires. Un carcinome basocellulaire peut envahir toute la paupière et se propager vers le cul-de-sac puis vers la conjonctive bulbaire.
[1] Shields CL et al. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004 ; 111 : 1747-54.
[2] Kenawy N et al. Conjunctival melanoma and melanocytic intra-epithelial neoplasia. Eye (Lond) 2013 ; 27 : 142-52.
[3] Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol 1997 ; 115 : 808-15.
[4] Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature 1968 ; 218 : 652-6.
[5] Robbins JH et al. Xeroderma pigmentosum. An inherited diseases with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med 1974 ; 80 : 221-48.
[6] Fazaa B, Kamoun MR. Xeroderma pigmentosum. Ann Dermatol Venereol 2003 ; 130 : 69-73.
[7] Kraemer KH, Slor H. Xeroderma pigmentosum. Clin Dermatol 1985 ; 3 : 33-69.
[8] Stary A, Sarasin A. Xeroderma pigmentosum. Presse Med 1997 ; 26 : 1992-7.
[9] Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 1987 ; 123 : 241-50.
[10] Alfawaz AM, Al-Hussain HM. Ocular manifestations of xeroderma pigmentosum at a tertiary eye care center in Saudi Arabia. Ophthal Plast Reconstr Surg 2011 ; 27 : 401-4.
[11] Brooks BP et al. Ocular manifestations of xeroderma pigmentosum : long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology 2013 ; 120 : 1324-36.
[12] Gupta N, Sachdev R, Tandon R. Ocular surface squamous neoplasia in xeroderma pigmentosum : clinical spectrum and outcome. Graefes Arch Clin Exp Ophthalmol 2011 ; 249 : 1217-21.
[13] Kaliki S et al. Conjunctival papilloma : features and outcomes based on age at initial examination. JAMA Ophthalmol 2013 ; 131 : 585-93.
[14] Minchiotti S et al. Conjunctival papilloma and human papillomavirus : identification of HPV types by PCR. Eur J Ophthalmol 2006 ; 16 : 473-7.
[15] Kothari M, Mody K, Chatterjee D. Resolution of recurrent conjunctival papilloma after topical and intralesional interferon alpha2b with partial excision in a child. J Aapos 2009 ; 13 : 523-5.
[16] Kalogeropoulos C et al. Squamous cell papilloma of the conjunctiva due to human papillomavirus (HPV) : presentation of two cases and review of literature. Clin Ophthalmol 2012 ; 6 : 1553-61.
[17] Chang SW, Huang ZL. Oral cimetidine adjuvant therapy for recalcitrant, diffuse conjunctival papillomatosis. Cornea 2006 ; 25 : 687-90.
[18] Shields CL et al. Oral cimetidine (Tagamet) for recalcitrant, diffuse conjunctival papillomatosis. Am J Ophthalmol 1999 ; 128 : 362-4.
[19] Ostergaard J, Prause JU, Heegaard S. Caruncular lesions in Denmark 1978-2002 : a histopathological study with correlation to clinical referral diagnosis. Acta Ophthalmol Scand 2006 ; 84 : 130-6.
[20] Khong JJ et al. Sebaceous gland carcinoma of the eyelid presenting as a conjunctival papilloma. Clin Experiment Ophthalmol 2005 ; 33 : 197-8.
[21] Rudkin AK, Dodd T, Muecke JS. The differential diagnosis of localised amelanotic limbal lesions : a review of 162 consecutive excisions. Br J Ophthalmol 2011 ; 95 : 350-4.
[22] Sjo NC, et al. Human papillomavirus in normal conjunctival tissue and in conjunctival papilloma : types and frequencies in a large series. Br J Ophthalmol 2007 ; 91 : 1014-5.
[23] Bredow L et al. Recurrent conjunctival papilloma progressing into squamous cell carcinoma with change of HPV-finding during the course. Br J Ophthalmol 2009 ; 93 : 1437, 1451.
[24] Kaeser PF et al. Tumors of the caruncle : a clinicopathologic correlation. Am J Ophthalmol 2006 ; 142 : 448-55.
[25] Oellers P et al. Conjunctival keratoacanthoma. Br J Ophthalmol 2014 ; 98 : 275-6.
[26] Cai R et al. Clinicopathological features of a suspected case of hereditary benign intraepithelial dyskeratosis with bilateral corneas involved : a case report and mini review. Cornea 2011 ; 30 : 1481-4.
[27] Ferrari G, Vigano M. Images in clinical medicine. Bitot’s spot in vitamin A deficiency. N Engl J Med 2013 ; 368 : e29.
[28] Tseng SH et al. Conjunctival seborrheic keratosis. Ophthalmology 2010 ; 117 : 190-190.e2.
[29] Mortemousque B et al. Actinic keratosis of the conjunctiva. Apropos of a clinical case. J Fr Ophtalmol 1998 ; 21 : 458-61.
[30] Mittal R, Meena M, Saha D. Actinic granuloma of the conjunctiva in young women. Ophthalmology 2013 ; 120 : 1786-9.
[31] Gallagher MJ et al. Actinic granuloma of the conjunctiva. Br J Ophthalmol 2003 ; 87 : 1044-5.
[32] Amoli FA, Heidari AB. Survey of 447 patients with conjunctival neoplastic lesions in Farabi Eye Hospital, Tehran, Iran. Ophthalmic Epidemiol 2006 ; 13 : 275-9.
[33] Lee GA, Hirst LW, Sheehan M. Knowledge of sunlight effects on the eyes and protective behaviors in the general community. Ophthalmic Epidemiol 1994 ; 1 : 67-84.
[34] Kiire CA, Dhillon B. The aetiology and associations of conjunctival intraepithelial neoplasia. Br J Ophthalmol 2006 ; 90 : 109-13.
[35] Alves LF et al. Expression of SIRT1 in ocular surface squamous neoplasia. Cornea 2012 ; 31 : 817-9.
[36] Woods M et al. Detecting human papillomavirus in ocular surface diseases. Invest Ophthalmol Vis Sci 2013 ; 54 : 8069-78.
[37] Oellers P et al. Prevalence, treatment, and outcomes of coexistent ocular surface squamous neoplasia and pterygium. Ophthalmology 2013 ; 120 : 445-50.
[38] Kao AA et al. Clinicopathologic correlation of ocular surface squamous neoplasms at Bascom Palmer Eye Institute : 2001 to 2010. Ophthalmology 2012 ; 119 : 1773-6.
[39] Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol 1997 ; 115 : 808-15.
[40] Erie JC, Campbell RJ, Liesegang TJ. Conjunctival and corneal intraepithelial and invasive neoplasia. Ophthalmology 1986 ; 93 : 176-83.
[41] Galor A et al. Predictors of ocular surface squamous neoplasia recurrence after excisional surgery. Ophthalmology 2012 ; 119 : 1974-81.
[42] Peksayar G, Soyturk MK, Demiryont M. Long-term results of cryotherapy on malignant epithelial tumors of the conjunctiva. Am J Ophthalmol 1989 ; 107 : 337-40.
[43] Kim JW, Abramson DH. Topical treatment options for conjunctival neoplasms. Clin Ophthalmol 2008 ; 2 : 503-15.
[44] Frucht-Pery J et al. Mitomycin C treatment for conjunctival-corneal intraepithelial neoplasia : a multicenter experience. Ophthalmology 1997 ; 104 : 2085-93.
[45] Hirst LW. Randomized controlled trial of topical mitomycin C for ocular surface squamous neoplasia : early resolution. Ophthalmology 2007 ; 114 : 976-82.
[46] Zaki AA, Farid SF. Management of intraepithelial and invasive neoplasia of the cornea and conjunctiva : a long-term follow up. Cornea 2009 ; 28 : 986-8.
[47] Russell HC et al. Topical mitomycin C chemotherapy in the management of ocular surface neoplasia : a 10-year review of treatment outcomes and complications. Br J Ophthalmol 2010 ; 94 : 1316-21.
[48] Chen C et al. Mitomycin C as an adjunct in the treatment of localised ocular surface squamous neoplasia. Br J Ophthalmol 2004 ; 88 : 17-8.
[49] Birkholz ES et al. Treatment of ocular surface squamous cell intraepithelial neoplasia with and without mitomycin C. Cornea 2011 ; 30 : 37-41.
[50] Sarici AM, Arvas S, Pazarli H. Combined excision, cryotherapy, and intraoperative mitomycin C (EXCRIM) for localized intraepithelial and squamous cell carcinoma of the conjunctiva. Graefes Arch Clin Exp Ophthalmol 2013 ; 251 : 2201-4.
[51] Lee JH et al. The effect of surgical wide excision and amniotic membrane transplantation with adjuvant topical mitomycin c treatment in recurrent conjunctival--corneal intraepithelial neoplasia. Semin Ophthalmol 2014 ; 29 : 192-5.
[52] Rozenman Y, Frucht-Pery J. Treatment of conjunctival intraepithelial neoplasia with topical drops of mitomycin C. Cornea 2000 ; 19 : 1-6.
[53] de Keizer RJ, de Wolff-Rouendaal D, Van Delft JL. Topical application of 5-fluorouracil in premalignant lesions of cornea, conjunctiva and eyelid. Doc Ophthalmol 1986 ; 64 : 31-42.
[54] Midena E et al. Treatment of conjunctival squamous cell carcinoma with topical 5-fluorouracil. Br J Ophthalmol 2000 ; 84 : 268-72.
[55] Parrozzani R et al. Topical 1 % 5-fluorouracil in ocular surface squamous neoplasia : a long-term safety study. Br J Ophthalmol 2011 ; 95 : 355-9.
[56] Maskin SL. Regression of limbal epithelial dysplasia with topical interferon. Arch Ophthalmol 1994 ; 112 : 1145-6.
[57] Karp CL, Moore JK, Rosa RH Jr. Treatment of conjunctival and corneal intraepithelial neoplasia with topical interferon alpha-2b. Ophthalmology 2001 ; 108 : 1093-8.
[58] Besley J, et al. Risk factors for ocular surface squamous neoplasia recurrence following treatment with topical mitomycin C and interferon alpha-2b. Am J Ophthalmol 2014 ; 157 : 287-93.
[59] Krilis M, Tsang H, Coroneo M. Treatment of conjunctival and corneal epithelial neoplasia with retinoic acid and topical interferon alfa-2b: long-term follow-up. Ophthalmology 2012 ; 119 : 1969-73.
[60] Huerva V, Mangues I. Treatment of conjunctival squamous neoplasias with interferon alpha 2ab. J Fr Ophtalmol 2008 ; 31 : 317-25.
[61] Schechter BA et al. Long-term follow-up of conjunctival and corneal intraepithelial neoplasia treated with topical interferon alfa-2b. Ophthalmology 2008 ; 115 : 1291-6, 1296.e1.
[62] Shah SU et al. Topical interferon alfa-2b for management of ocular surface squamous neoplasia in 23 cases : outcomes based on American Joint Committee on Cancer classification. Arch Ophthalmol 2012 ; 130 : 159-64.
[63] Shields CL et al. Interferon for ocular surface squamous neoplasia in 81 cases : outcomes based on the American Joint Committee on Cancer classification. Cornea 2013 ; 32 : 248-56.
[64] Newton R et al. Effect of ambient solar ultraviolet radiation on incidence of squamous-cell carcinoma of the eye. Lancet 1996 ; 347 : 1450-1.
[65] Carrilho C et al. Human papillomaviruses in intraepithelial neoplasia and squamous cell carcinoma of the conjunctiva : a study from Mozambique. Eur J Cancer Prev 2013 ; 22 : 566-8.
[66] Carreira H et al. HIV and HPV infections and ocular surface squamous neoplasia : systematic review and meta-analysis. Br J Cancer 2013 ; 109 : 1981-8.
[67] Chauhan S et al. Human papillomavirus : a predictor of better survival in ocular surface squamous neoplasia patients. Br J Ophthalmol 2012 ; 96 : 1517-21.
[68] Shields CL, Shields JA. Tumors of the conjunctiva and cornea. Surv Ophthalmol 2004 ; 49 : 3-24.
[69] Weinstein JE, Karp CL. Ocular surface neoplasias and human immunodeficiency virus infection. Curr Opin Infect Dis 2013 ; 26 : 58-65.
[70] Makupa II et al. Clinical factors associated with malignancy and HIV status in patients with ocular surface squamous neoplasia at Kilimanjaro Christian Medical Centre, Tanzania. Br J Ophthalmol 2012 ; 96 : 482-4.
[71] Shields CL et al. Conjunctival squamous cell carcinoma arising in immunosuppressed patients (organ transplant, human immunodeficiency virus infection). Ophthalmology 2011 ; 118 : 2133-2137.e1.
[72] Mukherjee B, Chatterjee R, Biswas J. Reverse masquerade syndrome : fungal adnexal infection mimicking carcinoma in a HIV-positive patient. Indian J Ophtalmol 2013 ; 61 : 521-3.
[73] Janin-Manificat H, et al. Conjunctival epidermoid carcinoma revealed by a chronic limbic corneal ulceration. J Fr Ophtalmol 2011 ; 34 : 108-12.
[74] Rootman DB, et al. Intraocular extension of conjunctival invasive squamous cell carcinoma after pterygium surgery and cataract extraction. Eye Contact Lens 2012 ; 38 : 133-6.
[75] Arend N, et al. Invasive squamous cell carcinoma of the conjunctiva. Ophthalmologe 2013 ; 110 : 57-60.
[76] Yousef YA, Finger PT. Squamous carcinoma and dysplasia of the conjunctiva and cornea : an analysis of 101 cases. Ophthalmology 2012 ; 119 : 233-40.
[77] Kivela T, Kujala E. Prognostication in eye cancer : the latest tumor, node, metastasis classification and beyond. Eye (Lond) 2013 ; 27 : 243-52.
[78] Mauriello JA Jr, Abdelsalam A, McLean IW. Adenoid squamous carcinoma of the conjunctiva--a clinicopathological study of 14 cases. Br J Ophthalmol 1997 ; 81 : 1001-5.
[79] Brownstein S. Mucoepidermoid carcinoma of the conjunctiva with intraocular invasion. Ophthalmology 1981 ; 88 : 1226-30.
[80] Alves LF, et al. Incidence of epithelial lesions of the conjunctiva in a review of 12,102 specimens in Canada (Quebec). Arq Bras Oftalmol 2011 ; 74 : 21-3.
[81] Lacour S, et al. Mucoepidermoid carcinoma of the conjunctiva with intraocular invasion. Apropos of a case. J Fr Ophtalmol 1991 ; 14 : 349-52.
[82] Jastrzebski A, et al. Histochemical analysis and immunohistochemical profile of mucoepidermoid carcinoma of the conjunctiva. Saudi J Ophthalmol 2012 ; 26 : 205-10.
[83] Shields CL, et al. Chemoreduction with topical mitomycin C prior to resection of extensive squamous cell carcinoma of the conjunctiva. Arch Ophthalmol 2005 ; 123 : 109-13.
[84] Maalouf TJ, et al. Sentinel lymph node biopsy in patients with conjunctival and eyelid cancers : experience in 17 patients. Ophthal Plast Reconstr Surg 2012 ; 28 : 30-4.
[85] Savar A, Esmaeli B. Re : « Sentinel lymph node biopsy in patients with conjunctival and eyelid cancers: experience in 17 patients ». Ophthal Plast Reconstr Surg 2012 ; 28 : 471, author reply 471-2.
[86] Pfeiffer, ML, Savar A, Esmaeli B. Sentinel lymph node biopsy for eyelid and conjunctival tumors: what have we learned in the past decade ? Ophthal Plast Reconstr Surg 2013 ; 29 : 57-62.
[87] Ramonas KM et al. Successful treatment of intraocularly invasive conjunctival squamous cell carcinoma with proton beam therapy. Arch Ophthalmol 2006 ; 124 : 126-8.
[88] Caujolle JP et al. Surgery and additional protontherapy for treatment of invasive and recurrent squamous cell carcinomas : technique and preliminary results. J Fr Ophtalmol 2009 ; 32 : 707-14.
[89] Graue GF, Tena LB, Finger PT. Electron beam radiation for conjunctival squamous carcinoma. Ophthal Plast Reconstr Surg 2011 ; 27 : 277-81.
[90] Sanchez-Perez JL, Fuentes-Sanchez C, Acosta-Acosta B. Conjunctival-corneal intraepithelial neoplasia (Bowen disease) treated with orthovoltage. Cornea 2011 ; 30 : 474-6.
[91] Stannard C et al. Radiotherapy for ocular tumours. Eye (Lond) 2013 ; 27 : 119-27.
[92] Aronow ME, Singh AD. Radiation therapy : conjunctival and eyelid tumors. Dev Ophthalmol 2013 ; 52 : 85-93.
[93] Robinson JW et al. Conjunctival mucoepidermoid carcinoma in a patient with ocular cicatricial pemphigoid and a review of the literature. Surv Ophthalmol 2006 ; 51 : 513-9.
[94] Husain SE et al. Primary basal cell carcinoma of the limbal conjunctiva. Ophthalmology 1993 ; 100 : 1720-2.
[95] Fino P et al. First reported case of primary basal cell carcinoma of the right caruncle: a case report and review of the literature. In Vivo 2013 ; 27 : 535-9.
[96] Taylor SF, Cook AE, Leatherbarrow B. Review of patients with basal cell nevus syndrome. Ophthal Plast Reconstr Surg 2006 ; 22 : 259-65.
C. Levy-Gabriel
Pour bon nombre d’entre nous, l’expression « tumeur mélanocytaire » est synonyme de lésion pigmentée, or les tumeurs mélanocytaires ne sont pas toujours pigmentées, les nævi comme les mélanomes peuvent se présenter sous une forme totalement achrome. À l’inverse, toutes les lésions pigmentées visualisables au niveau de la surface oculaire ne correspondent pas obligatoirement à des tumeurs mélanocytaires. Elles peuvent aussi correspondre à des dépôts de pigments intra- ou extracellulaires sans augmentation du nombre des mélanocytes (mélanoses réactionnelles ou secondaires, fig 11-10), ou être secondaires à une hypersécrétion de mélanine au sein de mélanocytes augmentés en nombre mais d’aspect normal (mélanose ethnique).
Les classifications et terminologies concernant les tumeurs mélanocytaires et autres lésions pigmentées de la surface oculaire sont nombreuses et parfois source de confusion. On trouve donc dans la littérature des classifications anciennes basées sur la clinique qui différencient les tumeurs congénitales des tumeurs acquises, ainsi que des classifications prenant en compte le potentiel évolutif de la lésion [1] et distinguant les lésions bénignes (mélanose ethnique, nævus, mélanose primitive acquise sans atypies) des lésions précancéreuses (mélanose primitive acquise avec atypies) et des tumeurs malignes (mélanome malin). Les classifications histologiques [2], plus récentes, ont le mérite de faire la différence entre les pigmentations réactionnelles, les hypermélanoses (mélanose ethnique) et les authentiques néoplasies mélanocytaires intra-épithéliales et stromales (nævus, mélanose primitive acquise sans et avec atypies, mélanome in situ et invasif). Le tableau 11-3 résume les différentes pathologies, avec leurs principales caractéristiques spécifiques, cliniques, évolutives et histologiques.
Nous détaillerons dans ce sous-chapitre les authentiques tumeurs mélanocytaires cornéoconjonctivales (nævus, mélanose primitive acquise sans et avec atypies, mélanome in situ et invasif), ainsi que les autres lésions mélanocytaires à évoquer devant un aspect pigmenté de la surface oculaire : la mélanose ethnique et la mélanocytose oculaire (prolifération mélanocytaire qui ne concerne pas la surface oculaire puisqu’elle est plus profonde, sclérale et uvéale mais souvent confondue avec une mélanose primitive acquise).

Fig. 11-10 Aspect de pigmentation réactionnelle sous-conjonctivale (mélanose réactionnelle) apparue plusieurs années après chirurgie sur strabisme (mise en place de fil d’argent).

Tableau 11-3 Résumé des différentes tumeurs mélanocytaires, avec leurs caractéristiques propres, cliniques, évolutives et histologiques.
Le nævus est la plus fréquente des tumeurs de la surface oculaire. Dans la série publiée par Shields, elle représente 28 % des tumeurs conjonctivales et 52 % des tumeurs mélanocytaires [3].
Le nævus est constitué de cellules næviques arrangées en thèques. Ces thèques sont initialement localisées à la jonction entre l’épithélium et le chorion. Au fur et à mesure de l’évolution, les thèques descendent dans le chorion et perdent leurs connexions avec l’épithélium. Un nævus localisé à la jonction épithélium–chorion est appelé nævus jonctionnel, alors qu’un nævus exclusivement localisé au niveau du chorion sera appelé nævus sous-épithélial ou intrastromal (fig 11-11 et 11-12). Lorsque la prolifération est à la fois jonctionnelle et sous-épithéliale, on parle de nævus composé (fig 11-13) [4].
Selon les auteurs, le nævus conjonctival est une tumeur congénitale classée parmi les hamartomes ou une tumeur acquise incluse dans les néoplasies. Il peut être présent cliniquement à la naissance ou apparaître au cours de la première ou deuxième décade. Il se présente sous la forme d’une lésion plane ou sessile avec un très discret relief, achrome avec aspect rosé dans 15 à 20 % des cas (fig 11-14), partiellement pigmenté dans 20 à 30 % (fig 11-15), et totalement pigmenté dans 50 à 65 % des cas (voir fig 11-13). La présence de kystes intralésionnels est fréquente – entre 60 % et 70 % des cas (voir fig 11-11 et 11-15). Dans environ un cas sur trois, on peut visualiser un fin réseau vasculaire au sein de la lésion (21 à 38 %) ou la présence de vaisseaux nourriciers dilatés (27 à 33 %) : voir fig 11-13a. La localisation de prédilection du nævus est la conjonctive bulbaire dans l’aire de la fente palpébrale (67 %). La localisation caronculaire ou au niveau du repli semi-lunaire est moins fréquente (31 %), et les nævi sont très rares au niveau de la conjonctive palpébrale (moins de 3 %) [5-7]. Le diamètre du nævus est en moyenne de 4 mm mais peut varier de 0,2 à 30 mm. Les lésions géantes (diamètre supérieur à 10 mm) représentent 5 % des cas [8]. En général stables dans le temps, les nævi peuvent cependant présenter quelques discrètes et très lentes modifications de la pigmentation ou de la taille (10 % des cas) [6, 7]. Pendant la puberté en particulier, la pigmentation peut augmenter et donner une fausse impression d’apparition récente ou de croissance. Le risque de transformation maligne est en réalité extrêmement faible, inférieur à 1 % [6, 9]. La meilleure attitude consiste donc en une surveillance régulière avec réalisation de photos comparatives. L’exérèse chirurgicale ne sera réalisée qu’en cas de modification objective et suspecte ou à la demande du patient pour des raisons esthétiques.

Fig. 11-11 Nævus kystique de la caroncule.
a. Aspect clinique. b. Aspect histologique. On observe une prolifération mélanocytaire constituée de mélanocytes réguliers, plus ou moins pigmentés, dépourvus d’atypies ou de mitose, organisés en plages ou en nids (thèques næviques) dans le chorion. Elle s’accompagne souvent de kystes d’inclusion épithéliale. Il n’y a pas d’envahissement de l’épithélium de surface.

Fig. 11-12 Nævus du repli semi-lunaire chez un enfant de 14 ans.
a. Aspect clinique. b. Aspect histologique de nævus intrastromal avec présence de petites thèques næviques composées de cellules mélanocytaires sans atypies, ni mitose au sein de la partie superficielle du chorion.

Fig. 11-13 Nævus composé chez un enfant mélanoderme de 16 ans.
a. Aspect clinique. b. Aspect histologique de nævus avec prolifération mélanocytaire sans atypies occupant la jonction entre le revêtement épithélial conjonctival et la partie superficielle et moyenne du chorion où elle s’organise en thèques ou en nappes.

Fig. 11-14 Aspect de nævus conjonctival achrome chez un enfant de 8 ans.

Fig. 11-15 Nævus kystique avec pigmentation hétérogène chez un homme de 65 ans.
La mélanocytose oculaire correspond en anatomie pathologique à une forme particulière de nævus : le nævus bleu. Les mélanocytes ont un aspect fusiforme identique à celui des cellules næviques du tractus uvéal et sont localisés à la partie profonde de l’épisclère et de la sclère, et au niveau du tractus uvéal ; la conjonctive (épithélium et chorion) n’est pas atteinte [4], il ne s’agit donc pas authentiquement d’une tumeur de la surface oculaire. Cliniquement, la mélanocytose oculaire se manifeste par une pigmentation sclérale bleu-gris souvent associée à une hétérochromie irienne (fig 11-16b et c). Elle est fréquemment confondue avec une prolifération mélanocytaire conjonctivale (nævus ou mélanose primitive acquise). Cette mélanocytose oculaire peut s’associer à une mélanocytose dermique avec présence d’un nævus bleu intéressant la zone cutanée péri-oculaire (fig 11-16a), on parle alors de mélanocytose oculodermique ou nævus d’Ota. Une mélanocytose orbitaire, méningée ou du palais mou peut coexister. L’atteinte est congénitale et unilatérale. Elle survient plus souvent chez des patients très pigmentés (par exemple sujets africains, hispaniques, asiatiques). Bien que rare chez les patients peu pigmentés, c’est dans cette population qu’il existe un risque de transformation maligne. Le mélanome apparaît alors en général au niveau du tractus uvéal (risque de 1/400 justifiant une surveillance annuelle du fond d’œil de ces patients) [10] ; les mélanomes cutanés, conjonctivaux, orbitaires ou méningés sont rares.

Fig. 11-16 Mélanocytose oculodermique ou nævus d’Ota.
Mélanocytoses : cutanée péri-oculaire (a), irienne (b) et oculaire avec aspect pigmenté grisâtre de la sclère (c).
La mélanose ethnique est relativement commune mais ne se voit que chez les patients à la peau pigmentée. En anatomie pathologique, elle se caractérise par une prolifération lentigineuse de mélanocytes d’aspect bénin le long de la membrane basale de l’épithélium. Cliniquement, la pigmentation conjonctivale est bilatérale, parfois un peu asymétrique. Elle est surtout visible au niveau du limbe sur 360° et au niveau de la conjonctive bulbaire (fig 11-17). L’atteinte des culs-de-sac ou de la conjonctive palpébrale est moins fréquente. La pigmentation est brune, plane, localisée à bords irréguliers ; elle peut parfois prendre un aspect très dense en petites mottes. Cette pigmentation est présente et connue du patient depuis sa jeunesse, elle est peu évolutive. Le risque de transformation maligne est extrêmement faible. La conduite à tenir se résume donc à une surveillance régulière.

Fig. 11-17 Mélanose ethnique chez un patient mélanoderme.
a. Œil droit. Noter le caractère plan de la pigmentation qui entoure le limbe sur 360°. b. Œil gauche. La mélanose ethnique est bilatérale avec parfois une petite asymétrie.
La mélanose primitive acquise est une lésion beaucoup moins fréquente que le nævus ou la mélanose ethnique. Elle se caractérise en anatomie pathologique par une prolifération anormale de mélanocytes au niveau de l’épithélium conjonctival ou cornéen. La prolifération est strictement intra-épithéliale, elle n’atteint pas la membrane basale et n’envahit pas le chorion.
Elle se différencie du nævus par un âge d’apparition plus tardif (entre 40 et 60 ans), une pigmentation irrégulière, toujours plane, non kystique ; la pigmentation peut s’étendre aux culs-de-sac, à la conjonctive palpébrale, ou au niveau de l’épithélium cornéen. Contrairement à la mélanocytose oculaire, la pigmentation est brune et non grisâtre, elle est située au niveau de la conjonctive et non pas au niveau de la sclère (fig 11-18). À la différence de la mélanose ethnique, elle apparaît chez des patients à la peau claire et l’atteinte est unilatérale. Dans certains cas, elle peut être achrome [11, 12].
La mélanose primitive acquise peut rester stable ou s’étendre progressivement sur une période de 10 ans ou plus. Le potentiel de dégénérescence en mélanome invasif est variable selon les cas. L’importance de ce risque est difficile à préciser sur le seul aspect clinique. Une étude rétrospective a cependant montré que l’étendue de l’atteinte conjonctivale (mesurée en nombre de quartiers horaires) pouvait être considérée comme un facteur de risque prédictif de transformation maligne : les lésions dont l’étendue est inférieure à un quadrant horaire ont un risque faible de transformation maligne, alors que les lésions de trois quadrants horaires et plus ont plus de 20 % de risque de transformation maligne [13]. Pour les lésions inférieures à un quadrant horaire, on pourrait donc se contenter d’une surveillance, alors qu’une biopsie devrait être envisagée pour les lésions de trois quadrants et plus.
En effet, la présence ou non d’atypies et leur importance sur l’analyse anatomo-pathologique permettent d’évaluer avec plus de précision le risque de dégénérescence maligne [12, 14]. On distingue ainsi la mélanose primitive acquise sans atypies, qui est relativement similaire à la mélanose ethnique en histologie, avec une prolifération lentigineuse de mélanocytes sans caractère atypique, limitée au niveau de la membrane basale de l’épithélium (fig 11-19). Cette mélanose primitive acquise sans atypies n’a pas de potentiel évolutif vers le mélanome. À l’opposé, la mélanose primitive acquise avec atypies se caractérise en histologie par :
une migration de mélanocytes au niveau de l’épithélium superficiel (ascension pagétoïde) avec discohésion ;
un degré variable de pléomorphisme cellulaire, certains groupes cellulaires allant jusqu’à présenter une morphologie épithélioïde, avec un volumineux noyau hyperchromatique et un important nucléole ;
des figures mitotiques pouvant être présentes ;
une réponse inflammatoire chronique pouvant être notée au niveau du chorion.
La prolifération de mélanocytes atypiques peut rester confinée au niveau de la membrane basale ou intéresser une épaisseur plus ou moins importante de l’épithélium [4]. En fonction de l’importance de ces anomalies, on peut classifier ces lésions en prolifération mélanocytaires intra-épithéliales avec atypies minimes, modérées (fig 11-20) ou sévères (fig 11-21) [2]. Dans les mélanoses primitives acquises avec atypies minimes, on n’a que quelques mélanocytes au niveau de l’épithélium superficiel et le risque de transformation maligne, s’il existe, est minime. En cas d’atypies modérées ou plus importantes, le risque de transformation maligne est corrélé au degré d’atypies [12, 14]. En cas de lésion envahissant toute l’épaisseur de l’épithélium avec des atypies sévères, on parlera de mélanome in situ. Cette classification des proliférations mélanocytaires intra-épithéliales en fonction des atypies en trois classes (minimes, modérées ou sévères) est cependant imprécise et par conséquent peu objective et reproductible. Damato et al. ont donc proposé un système de score de 0 à 10 calculé en fonction :
de l’aspect de l’extension horizontale (basale, pagétoïde ou nodulaire) ;
du degré d’extension verticale : limité aux couches basales ; inférieur à 50 % de l’épaisseur de l’épithélium ; de 50 à 90 % de l’épaisseur ; supérieur à 90 % de l’épaisseur ;
de l’importance des atypies cytonucléaires (taille du noyau, abondance du cytoplasme, importance des mitoses) [2].
Un score à 0 correspond à une simple mélanose ethnique ou primary acquired melanosis (PAM) sans atypies, un score à 1 équivaut à une PAM avec atypies minimes, un score à 2-3 à une PAM avec atypies modérées, un score à 4 à une PAM avec atypies sévères et au-delà de 5, la lésion sera considérée comme un mélanome in situ [11].
La mélanose primitive acquise avec atypies est similaire sur le plan histologique à la mélanose de Dubreuilh au niveau cutané. Il est à noter qu’une mélanose de Dubreuilh cutanée palpébrale peut parfois être présente en continuité avec une mélanose primitive acquise de la conjonctive palpébrale.
Les mélanoses primitives acquises sans atypies ne nécessitent qu’une simple surveillance. En cas de mélanose primitive acquise avec atypies, le traitement a pour but d’éviter l’apparition d’un mélanome invasif. Il n’y a pas actuellement de consensus concernant la meilleure prise en charge. Pour certains auteurs, en cas de lésions peu étendues (inférieures à un quadrant horaire), on peut se contenter d’une surveillance. En cas de lésions plus étendues ou évolutives, la classique exérèse chirurgicale (avec ou sans greffe de membrane amniotique), associée à une cryo-application, a actuellement tendance à être remplacée par des biopsies (éventuellement multiples) associées à une chimiothérapie topique. Ces chimiothérapies topiques présentent l’avantage de traiter l’ensemble de la conjonctive, y compris les zones d’infiltrations mélanocytaires intra-épithéliales achromes non visibles cliniquement ; par contre, elles sont inefficaces sur les proliférations sous-épithéliales. Elles représentent donc une bonne alternative à l’exérèse chirurgicale en cas de mélanose primitive acquise avec atypies, en particulier en cas d’atteinte diffuse ou multifocale. La molécule la plus utilisée dans ce contexte est la mitomycine C (MMC), les séries publiées rapportant avec des protocoles variables (concentrations, durées et nombre de cycles) une diminution ou une disparition complète de la pigmentation conjonctivale [15, 16]. Les complications de la MMC collyre sont en général passagères (hyperémie conjonctivale, chémosis, kératite ponctuée superficielle). Une série a cependant rapporté des effets secondaires à long terme voire définitifs : larmoiement, insuffisance limbique [17]. L’importance de la dose cumulative de MMC (concentration à 0,04 %, cycles longs et nombre importants de cycles), ainsi que les antécédents d’exérèses chirurgicales larges avec cryo-application pourraient pour certains auteurs favoriser la survenue d’une insuffisance limbique [17, 18]. Le traitement des mélanoses primitives acquises avec atypies par interféron alpha-2b en collyre n’a fait l’objet que d’une publication rapportant une diminution ou une disparition de la pigmentation dans sept cas sur neuf sans aucun effet secondaire [19].

Fig. 11-18 Aspect clinique d’une mélanose primitive acquise ou prolifération mélanocytaire intra-épithéliale.

Fig. 11-19 Prolifération mélanocytaire intra-épithéliale sans atypies.
Noter la prolifération lentigineuse des mélanocytes cantonnée au niveau de la membrane basale, avec des intervalles d’épithélium sain. Il n’y a ni atypies, ni mitose, ni ascension pagétoïde.
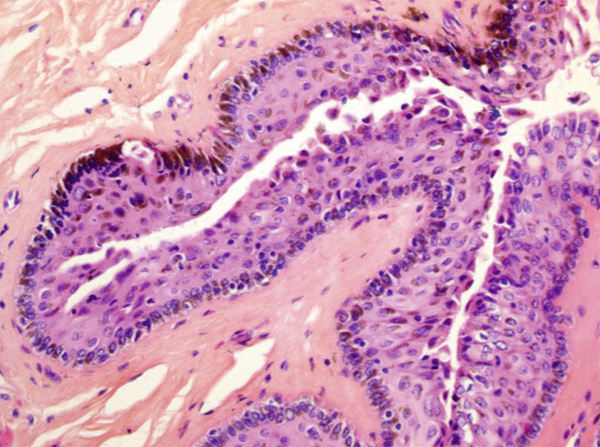
Fig. 11-20 Prolifération mélanocytaire intra-épithéliale avec atypies modérées.
Noter l’aspect partiellement désorganisé des couches basales et parabasales de l’épithélium. Au niveau des assises plus superficielles, l’architecture du corps muqueux de Malpighi est respectée et on n’observe pas d’aspect d’ascension d’éléments mélanocytaires.

Fig. 11-21 Prolifération mélanocytaire intra-épithéliale intéressant plus de 90 % de l’épaisseur de l’épithélium conjonctival avec atypies sévères et pouvant être considérée comme un mélanome in situ.
Le mélanome conjonctival est une tumeur maligne potentiellement létale. C’est une tumeur rare, elle ne représente que 5 % des mélanomes oculaires primitifs [20]. Son incidence est en augmentation [21]. Elle a été évaluée aux États-Unis et en Finlande à respectivement 0,54 et 0,80 pour 1 000 000 d’habitants par an à la fin des années 1990, soit environ 2 fois plus que 30 ans auparavant [22, 23].
Le mélanome conjonctival est caractérisé en anatomie pathologique par une prolifération tumorale de mélanocytes atypiques qui intéressent l’épithélium conjonctival, franchissent la membrane basale et envahissent le chorion sous-jacent (fig 11-22b). L’accès à la vascularisation présente au niveau du chorion (en particulier la vascularisation lymphatique) explique le risque de dissémination métastatique et de décès du patient (entre 16 et 32 % 5 ans) [24-26].
La morphologie cellulaire du mélanome est variable, de fusiforme à épithélioïde. Les lésions les plus agressives présentent des figures mitotiques. Le diagnostic basé uniquement sur les caractéristiques histologiques peut être parfois difficile. L’immuno-histochimie aide alors en utilisant différents marqueurs. Les colorations immuno-histochimiques des mélanocytes, comme le rouge Melan-A, MART-1, HMB-45, S100, peuvent être utiles pour identifier l’origine mélanocytaire des cas problématiques. S100 est sensible mais non spécifique du mélanome conjonctival, alors que HMB-45 est moins sensible mais plus spécifique.
Le terrain de prédilection est l’adulte à la peau claire d’environ 60 ans. Les cas rapportés chez l’enfant de moins de 15 ans sont exceptionnels (moins de 28 cas dans la littérature dont seulement huit bien documentés) [27].
Dans 50 à 70 % des cas, le mélanome conjonctival se développe sur une mélanose conjonctivale primitive acquise avec atypies (fig 11-23a et 11-24), mais il peut aussi apparaître sur une conjonctive saine – mélanome « de novo » (fig 11-25 et 11-26a) ou, plus rarement, provenir de la dégénérescence d’un nævus conjonctival préexistant [28, 29].
L’aspect clinique du mélanome est variable. Il se présente en général sous la forme d’une lésion pigmentée en relief (voir fig 11-25), plus ou moins nodulaire et vascularisée, mais il peut aussi être achrome, même s’il survient sur une mélanose primitive acquise avec atypies pigmentées (voir fig 11-23). Il peut intéresser n’importe quelle portion de la conjonctive (limbique, bulbaire, cul-de-sac ou conjonctive palpébrale), s’étendre en surface au niveau de la cornée ; l’atteinte peut être plurifocale.
La différenciation clinique entre une prolifération mélanocytaire intra-épithéliale avec atypies sévères, un mélanome in situ et un mélanome invasif débutant peut parfois être très difficile. De même, un mélanome achrome peut poser des problèmes de diagnostic différentiel avec un carcinome épidermoïde ou avec des lésions bénignes à présentation pseudo-tumorale (granulome inflammatoire, ptérygion, etc.). Le contexte (terrain, âge, ancienneté et évolutivité de la lésion) permet d’orienter le diagnostic. Les images en OCT (optical coherence tomography) à haute résolution peuvent aussi apporter une aide diagnostic intéressante et non invasive en permettant une bonne analyse de l’épithélium (épaisseur, réflectivité), et en différenciant les lésions intra-épithéliales des lésions sous-épithéliales. Une étude prospective a montré récemment une bonne corrélation entre les caractéristiques des images OCT et le diagnostic histologique [30].
Le traitement du mélanome conjonctival comprend en premier lieu l’exérèse chirurgicale de la lésion macroscopique. Pour certains auteurs, les biopsies doivent être évitées car elles seraient susceptibles de disséminer des cellules tumorales à distance du site initial [31]. Pour les mêmes raisons, toute infiltration sous-conjonctivale est à proscrire (anesthésiques locaux). Une « no touch » technique est recommandée : manipulation minimum de la tumeur ; utilisation d’alcool absolu pour décoller l’épithélium cornéen ; cautérisation des vaisseaux nourriciers ; exérèse de la tumeur en bloc avec marges latérales de quelques millimètres ; changements d’instruments pour la reconstruction [11, 32]. L’intérêt des kératectomies ou sclérectomies lamellaires dans le but d’obtenir une exérèse complète reste discuté, cette technique pouvant favoriser les récidives profondes et ne prémunissant pas des risques de récidive locale [31]. En cas de lésions très étendues ou multifocales, la réalisation d’une exentération orbitaire peut être nécessaire, mais l’intérêt de cette chirurgie dans la diminution du risque de dissémination métastatique n’a jamais été prouvé [25, 33].
Plusieurs études ont montré que l’absence de traitement adjuvant après exérèse chirurgicale augmentait le risque de récidive locale [31, 34]. Les deux principaux traitements adjuvants utilisés sont la cryo-application et la radiothérapie complémentaire. La réalisation d’une double cryo-application des berges d’exérèse et du lit tumoral après exérèse chirurgicale réduit significativement le risque de récidive locale, comparé à l’exérèse chirurgicale seule [35].
Concernant la radiothérapie complémentaire, la curiethérapie (au strontium 90, au rhuténium ou à l’iode 125) permet de réduire le risque de récidive locale avec des effets secondaires acceptables (voir fig 11-22) [31, 36, 37]. Pour certains auteurs, ces techniques peuvent même être utilisées lorsque la tumeur présente une localisation palpébrale [38] ou envahit la cornée ou la sclère [39]. La protonthérapie, technique d’irradiation homogène et ultraprécise, donne aussi de très bons résultats (fig 11-26 et 11-27) [40]. Dans notre expérience à l’Institut Curie portant sur 62 premières localisations de mélanomes conjonctivaux irradiés par protons, le taux de récidive locale à 5 ans est de 19,7 %. Au niveau des séquelles radiques, on note essentiellement le développement d’une cataracte (constatation chez 12 patients). L’indication à une irradiation complémentaire est en général retenue lorsque l’exérèse chirurgicale est incomplète, macroscopiquement ou sur l’analyse anatomo-pathologique (envahissement tumoral des berges latérales et/ou profondes). Mais ces dernières années, une irradiation complémentaire (curiethérapie ou protonthérapie) a été proposée systématiquement à tous les patients dans certains centres dont l’Institut Curie, l’analyse histologique des berges étant considérée comme peu fiable même lorsqu’elle est très rigoureuse [31, 36].
Dans la littérature, les facteurs pronostiques identifiés pour le risque de récidive locale concernent essentiellement la localisation tumorale, l’atteinte des culs-de-sac, de la caroncule ou de la conjonctive palpébrale étant défavorable [24, 25, 31]. Pour ce qui est du risque de dissémination métastatique et de mortalité, les facteurs pronostiques identifiés sont une épaisseur tumorale supérieure à 2 mm et une localisation tumorale défavorable (atteinte des culs-de-sac, caronculaire ou palpébrale) [24-26, 41]. La valeur pronostic de l’origine clinique ou histologique du mélanome (de novo, ou provenant d’une PAM) reste discutée [24, 29, 41]. Enfin, plusieurs études ont montré une forte corrélation entre la survenue de récidives locales et l’évolution métastatique [2, 24, 25, 42]. La classification clinique pronostique la plus récente est la 7e édition de la classification TNM de l’American Joint Committee on Cancer (AJCC) présentée dans le tableau 11-4. Elle a une bonne valeur prédictive en ce qui concerne le risque de récidive locale et de dissémination métastatique [43, 44].
Les taux de récidive locale et de mortalité rapportés dans la littérature varient selon les études et le type de traitement réalisé avec des taux à 5 ans entre 26 et 50 % pour les récidives locales et entre 7 et 32 % pour la mortalité [24-26, 33].
Le bilan d’extension des patients à risque métastatique comprend en général la palpation des aires ganglionnaires, une imagerie cervicale, thoracique, hépatique et parfois cérébrale, le type d’imagerie (échographie, scanner ou IRM) variant selon les équipes. L’intérêt de la biopsie du ganglion lymphatique sentinelle qui pour but d’identifier les patients présentant une micrométastase ganglionnaire infraclinique reste controversé [45]. Il n’y a pas actuellement de consensus sur quand et à qui le proposer. Et si 45 % des patients métastasent au niveau du ganglion lymphatique avant de développer des métastases viscérales, 50 % à l’opposé métastasent en premier lieu au niveau viscéral [26]. La place du PET scan corps entier reste encore mal définie. Il a été montré qu’il était supérieur à l’imagerie classique pour dépister les métastases systémiques, en particulier les atteintes osseuses, mais son utilisation reste limitée du fait de son coût élevé, des faux négatifs et de son manque de spécificité [46, 47].
Des travaux récents (utilisant multiplex ligation-dependent probe amplification) ont permis d’identifier une mutation BRAF V600E dans 50 % des cas de mélanome conjonctival primitif et dans plus de 50 % des lésions métastatiques et ont donc conduit à évaluer, par analogie aux cas de mélanomes cutanés avec mutation BRAF V600E, l’efficacité thérapeutique de l’inhibiteur BRAF PLX4023 (vemurafenib) spécifique de la mutation V600E dans le mélanome conjonctival [48].

Fig. 11-22 Mélanome invasif associé à une prolifération mélanocytaire intra-épithéliale avec atypies.
a. Aspect clinique avant traitement en 2005. b. Aspect histologique. Noter la prolifération mélanocytaire au niveau du chorion avec atypies cytonucléaires marquées et présence de mitoses. c. Aspect 3 ans après traitement par chirurgie et irradiation complémentaire par disque d’iode 125. Acuité visuelle 10/10.

Fig. 11-23 Mélanomes conjonctivaux achromes.

Fig. 11-24 Mélanome conjonctival pigmenté nodulaire au niveau du cul-de-sac inférieur apparu sur une prolifération mélanocytaire intra-épithéliale avec atypies très étendues.

Fig. 11-25 Mélanome de novo pigmenté en relief au niveau du limbe.

Fig. 11-26 Mélanome conjonctival de novo.
a. Avant traitement. b. Après exérèse chirurgicale, reconstruction par greffe de membrane amniotique et irradiation complémentaire par faisceaux de protons.

Fig. 11-27 Mélanome cornéoconjonctival apparu sur une mélanose primitive acquise.
a, b. Avril 2009 : aspect avant traitement. c, d. Mai 2009 : aspect après exérèse chirurgicale des lésions de mélanome invasif.
e, f. Octobre 2011 : 2 ans après irradiation complémentaire par faisceaux de protons et traitement de la prolifération intra-épithéliale résiduelle par mitomycine topique.

Tableau 11-4 Classification du mélanome conjonctival (d’après la 7e classification de l’American Joint Committee on Cancer).
a pTis : mélanome in situ (y compris la mélanose acquise primitive) avec atypie de l’épaisseur épithéliale normale supérieure à 75 %, avec caractéristiques cytologiques de cellules épithélioïdes, dont un cytoplasme abondant, des noyaux vésiculaires ou des nucléoles proéminents, ou la présence de réseaux intra-épithéliaux de cellules atypiques.
b Les quadrants correspondent à des quadrants horaires, commençant à partir du limbe (par exemple 6, 9, 12 et 3 heures) et allant de la cornée centrale jusqu’aux bords palpébraux et au-delà. Cela divise en deux la caroncule.
[1] Shields CL, Shields JA. Tumors of the conjunctiva and cornea. Surv Ophthalmol 2004 ; 49 : 3-24.
[2] Damato B, Coupland SE. Conjunctival melanoma and melanosis: a reappraisal of terminology, classification and staging. Clin Experiment Ophthalmol 2008 ; 36 : 786-95.
[3] Shields CL et al. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004 ; 111 : 1747-54.
[4] Desjardins L. Anatomie pathologique en ophtalmologie. Tumeurs intra-oculaires. Issy-les-Moulineaux : Elsevier Masson/SFO ; 2011-2012, p. 61.
[5] Gerner N et al. Conjunctival naevi in Denmark 1960–1980. A 21-year follow-up study. Acta Ophthalmol Scand 1996 ; 74 : 334-7.
[6] Shields CL et al. Conjunctival nevi : clinical features and natural course in 410 consecutive patients. Arch Ophthalmol 2004 ; 122 : 167-75.
[7] Levecq L, De Potter P, Jamart J. Conjunctival nevi clinical features and therapeutic outcomes. Ophthalmology 1016 ; 117 : 35-40.
[8] Shields CL et al. Giant conjunctival nevus : clinical features and natural course in 32 cases. JAMA Ophthalmol 2013 ; 131 : 857-63.
[9] Shields CL, Shields JA. Conjunctival tumors in children. Curr Opin Ophthalmol 2007 ; 18 : 351-60.
[10] Singh AD et al. Lifetime prevalence of uveal melanoma in white patients with oculo(dermal) melanocytosis. Ophthalmology 1998 ; 105 : 195-8.
[11] Kenawy N et al. Conjunctival melanoma and melanocytic intra-epithelial neoplasia. Eye (Lond) 2013 ; 27 : 142-52.
[12] Shields JA et al. Primary acquired melanosis of the conjunctiva : risks for progression to melanoma in 311 eyes. The 2006 Lorenz E. Zimmerman lecture. Ophthalmology 2008 ; 115 : 511-519.e2.
[13] Shields JA et al. Primary acquired melanosis of the conjunctiva : experience with 311 eyes. Trans Am Ophthalmol Soc 2007 ; 105 : 61-71, discussion 71-2.
[14] Sugiura M et al. Low-risk and high-risk histologic features in conjunctival primary acquired melanosis with atypia: Clinicopathologic analysis of 29 cases. Am J Surg Pathol 2007 ; 31 : 185-92.
[15] Kurli M, Finger PT. Topical mitomycin chemotherapy for conjunctival malignant melanoma and primary acquired melanosis with atypia : 12 years’ experience. Graefes Arch Clin Exp Ophthalmol 2005 ; 243 : 1108-14.
[16] Pe’er J, Frucht-Pery J. The treatment of primary acquired melanosis (PAM) with atypia by topical Mitomycin C. Am J Ophthalmol 2005 ; 139 : 229-34.
[17] Ditta LC, Shildkrot Y, Wilson MW. Outcomes in 15 patients with conjunctival melanoma treated with adjuvant topical mitomycin C : complications and recurrences. Ophthalmology 2011 ; 118 : 1754-9.
[18] Khong JJ, Muecke J. Complications of mitomycin C therapy in 100 eyes with ocular surface neoplasia. Br J Ophthalmol 2006 ; 90 : 819-22.
[19] Herold TR, Hintschich C. Interferon alpha for the treatment of melanocytic conjunctival lesions. Graefes Arch Clin Exp Ophthalmol 2010 ; 248 : 111-5.
[20] Isager P et al. Uveal and conjunctival malignant melanoma in Denmark, 1943-97 : incidence and validation study. Ophthalmic Epidemiol 2005 ; 12 : 223-32.
[21] Triay E et al. Time trends in the incidence of conjunctival melanoma in Sweden. Br J Ophthalmol 2009 ; 93 : 1524-8.
[22] Yu GP et al. Conjunctival melanoma : is it increasing in the United States ? Am J Ophthalmol 2003 ; 135 : 800-6.
[23] Tuomaala S, Kivela T. Conjunctival melanoma : is it increasing in the United States ? Am J Ophthalmol 2003 ; 136 : 1189-90, author reply 1190.
[24] Anastassiou G et al. Prognostic value of clinical and histopathological parameters in conjunctival melanomas : a retrospective study. Br J Ophthalmol 2002 ; 86 : 163-7.
[25] Shields CL et al. Conjunctival melanoma : risk factors for recurrence, exenteration, metastasis, and death in 150 consecutive patients. Arch Ophthalmol 2000 ; 118 : 1497-507.
[26] Tuomaala S, Kivela T. Metastatic pattern and survival in disseminated conjunctival melanoma: implications for sentinel lymph node biopsy. Ophthalmology 2004 ; 111 : 816-21.
[27] Taban M, Traboulsi EI. Malignant melanoma of the conjunctiva in children : a review of the international literature 1965-2006. J Pediatr Ophthalmol Strabismus 2007 ; 44 : 277-82, quiz 298-9.
[28] Brownstein S. Malignant melanoma of the conjunctiva. Cancer Control 2004 ; 11 : 310-6.
[29] Shields CL et al. Conjunctival melanoma: outcomes based on tumor origin in 382 consecutive cases. Ophthalmology 2011 ; 118 : 389-95.
[30] Shousha MA et al. Diagnosis of ocular surface lesions using ultra-high-resolution optical coherence tomography. Ophthalmology 2013 ; 120 : 883-91.
[31] Damato B, Coupland SE. An audit of conjunctival melanoma treatment in Liverpool. Eye (Lond) 2009 ; 23 : 801-9.
[32] Shields JA, Shields CL, De Potter P. Surgical management of conjunctival tumors. The 1994 Lynn B. McMahan Lecture. Arch Ophthalmol 1997 ; 115 : 808-15.
[33] Paridaens AD et al. Orbital exenteration in 95 cases of primary conjunctival malignant melanoma. Br J Ophthalmol 1994 ; 78 : 520-8.
[34] Werschnik C, Lommatzsch PK. Long-term follow-up of patients with conjunctival melanoma. Am J Clin Oncol 2002 ; 25 : 248-55.
[35] De Potter P et al. Clinical predictive factors for development of recurrence and metastasis in conjunctival melanoma : a review of 68 cases. Br J Ophthalmol 1993 ; 77 : 624-30.
[36] Karim R, Conway RM. Conservative resection and adjuvant plaque brachytherapy for early-stage conjunctival melanoma. Clin Experiment Ophthalmol 2011 ; 39 : 293-8.
[37] Missotten GS et al. Conjunctival melanoma in the Netherlands : a nationwide study. Invest Ophthalmol Vis Sci 2005 ; 46 : 75-82.
[38] Stannard CE et al. Malignant melanoma of the eyelid and palpebral conjunctiva treated with iodine-125 brachytherapy. Ophthalmology 2000 ; 107 : 951-8.
[39] Walsh-Conway N, Conway RM. Plaque brachytherapy for the management of ocular surface malignancies with corneoscleral invasion. Clin Experiment Ophthalmol 2009 ; 37 : 577-83.
[40] Wuestemeyer H et al. Proton radiotherapy as an alternative to exenteration in the management of extended conjunctival melanoma. Graefes Arch Clin Exp Ophthalmol 2006 ; 244 : 438-46.
[41] Paridaens AD et al. Prognostic factors in primary malignant melanoma of the conjunctiva: a clinicopathological study of 256 cases. Br J Ophthalmol 1994 ; 78 : 252-9.
[42] Desjardins L et al. Prognostic factors in malignant melanoma of the conjunctiva. An anatomo-clinical study of 56 patients. J Fr Ophtalmol 1999 ; 22 : 315-21.
[43] Yousef YA, Finger PT. Predictive value of the seventh edition American Joint Committee on Cancer staging system for conjunctival melanoma. Arch Ophthalmol 2012 ; 130 : 599-606.
[44] Shields CL et al. American Joint Committee on Cancer (AJCC) clinical classification predicts conjunctival melanoma outcomes. Ophthal Plast Reconstr Surg 1097 ; 28 : 313-23.
[45] Cohen VM et al. Prospective study of sentinel lymph node biopsy for conjunctival melanoma. Br J Ophthalmol2013 ; 97 : 1525-9.
[46] Kurli M, Chin K, Finger PT. Whole-body 18 FDG PET/CT imaging for lymph node and metastatic staging of conjunctival melanoma. Br J Ophthalmol 2008 ; 92 : 479-82.
[47] Esmaeli B. Regional lymph node assessment for conjunctival melanoma : sentinel lymph node biopsy and positron emission tomography. Br J Ophthalmol 2008 ; 92 : 443-5.
[48] Lake SL et al. Multiplex ligation-dependent probe amplification of conjunctival melanoma reveals common BRAF V600E gene mutation and gene copy number changes. Invest Ophthalmol Vis Sci 2011 ; 52 : 5598-604.
N. Cassoux
Les maladies hématologiques, en particulier les lymphomes, peuvent atteindre l’œil sous différentes formes. Ce que l’on appelle les lymphomes oculaires est un fourre-tout regroupant les atteintes intra-oculaires et les atteintes des annexes de l’œil et de l’orbite. Les atteintes intra-oculaires ont fait l’objet récemment d’une nouvelle classification séparant les lymphomes vitréorétiniens (vitréen, vitréorétinien et rétinien), les lymphomes choroïdiens et les lymphomes de l’iris. L’atteinte intra-oculaire la plus fréquente est liée au lymphome oculaire et cérébral primitif. À côté de ces atteintes intra-oculaires, il existe une atteinte des annexes de l’œil (conjonctive, glande lacrymale) et de l’orbite dont la plus fréquente est liée au lymphome de type MALT (mucosa-associated lymphoid tissue) [1]. Le lymphome peut atteindre l’œil de façon directe (infiltration des tissus de l’œil par les cellules lymphomateuses) ou de façon indirecte « paranéoplasique », comme pour la maladie de Hodgkin, qui peut donner exceptionnellement une inflammation de l’œil (épisclérite, sclérite, vascularite rétinienne, uvéite, papillopathie inflammatoire) réactionnelle de mécanisme mal compris [2, 3].
Beaucoup plus rarement, les autres hémopathies peuvent induire des atteintes oculaires soit métastatiques (par exemple infiltration de l’uvée par les cellules leucémiques) soit par accumulation d’immunoglobulines dans la cornée comme dans le myélome, soit par un syndrome d’hyperviscosité.
Dans ce sous-chapitre, seules les atteintes de la conjonctive et des annexes les plus fréquentes seront détaillées.
Ce sont des proliférations monoclonales malignes développées à partir des cellules lymphoïdes B ou T de degré de maturation variable, à point de départ périphérique. Ils sont caractérisés par une très grande diversité clinico-biologique, décrits dans la dernière classification de l’Organisation mondiale de la santé (tableau 11-5), et de pronostic très différent [4].

Tableau 11-5 Classification OMS des lymphomes (2008).
ALK : anaplastic lymphoma kinase ; EBV : Epstein-Barr virus ; HHV : human herpesvirus ; HTLV-1 : virus T-lymphotrope 1 humain ; SNC : système nerveux central.
(Source : Swerdlow SH, Campo E, Harris NL, et al. Eds. WHO Classification of tumours of haematopoietic and lymphoid tissues. Lyon : IARC ; 2008.)
Les lymphomes non hodgkiniens (LNH) sont les plus fréquentes des hémopathies malignes. Cette incidence a doublé en 20 ans [5]. Ils atteignent un peu plus souvent les hommes que les femmes (sex-ratio : 1,3:1). Environ 10 000 nouveaux cas sont diagnostiqués par an en France. Chez l’adulte, la médiane d’âge se situe autour de 60 ans. Les LNH sont parmi les affections cancéreuses les plus fréquentes de l’enfant, l’adolescent et l’adulte jeune. Les LNH de type B sont les plus nombreux, sauf au Japon où les lymphomes de type T sont majoritaires. Chez l’adulte, les lymphomes les plus fréquents sont les LNH diffus à grandes cellules B (33 %) et les LNH folliculaires (22 %).
Les manifestations cliniques sont variées. Il peut s’agir de la découverte par le patient d’une adénopathie périphérique, ou de la manifestation d’une atteinte ganglionnaire disséminée et et/ou multiviscérale. L’évolution peut être lente ou rapidement évolutive. Des signes généraux tels qu’une fièvre, un amaigrissement ou des sueurs profuses peuvent être révélateurs.
Le diagnostic repose sur l’analyse histologique d’une adénopathie ou d’une atteinte viscérale en l’absence d’adénopathie. L’analyse histologique est complétée d’une analyse immuno-histochimique. Des analyses complémentaires en cytogénétique (caryotype), cytogénétique moléculaire (fluorescence in situ hybridization ou FISH) et biologie moléculaire sont nécessaires pour la caractérisation de certaines entités (voir tableau 11-5).
Aucun facteur déclenchant des LNH n’est actuellement reconnu.
1. Certaines situations pathologiques peuvent favoriser l’apparition d’un LNH :
les déficits immunitaires et les allogreffes d’organe avec immunosuppression ;
les maladies dysimmunitaires (syndrome de Gougerot-Sjögren, thyroïdite d’Hashimoto) ;
l’infection par le virus de l’immunodéficience humaine (VIH).
2. Certains LNH peuvent être induits par un virus :
le lymphome de Burkitt endémique africain lié au virus d’Epstein-Barr (EBV) [6] ;
certains lymphomes T liés au virus T-lymphotrope 1 humain (HTLV-1) ;
les lymphomes des immunodéprimés et EBV ;
le virus de l’hépatite C associé à certains lymphomes de bas grade histologique [7].
3. La stimulation antigénique chronique induite par Helicobacter pylori est un facteur favorisant la survenue du lymphome de type MALT gastrique [8].
Une fois le diagnostic histologique établi, le bilan initial doit permettre d’évaluer l’ensemble des aires ganglionnaires et/ou atteintes extraganglionnaires atteintes. Il comprend au minimum :
tomodensitométrie (TDM) thoraco-abdomino-pelvien ± cervical ;
positon emission tomography scan (PET scan) corps entier (pas pour certaines histologies) ;
biopsie ostéomédullaire ;
ponction lombaire pour les LNH agressifs.
D’autres explorations seront guidées par les points d’appel cliniques : imagerie par résonance magnétique (IRM) cérébrale, échographie testiculaire, exploration digestive, explorations osseuses, examen ophtalmologique, etc.
Ce bilan permet de définir le stade selon la classification d’Ann Arbor (tableau 11-6).
Les atteintes extraganglionnaires peuvent atteindre la sphère ORL, le tractus digestif, le foie, la glande mammaire, les gonades, les os, la peau, la moelle osseuse et le système neurologique périphérique et central y compris l’œil (vitré et nerf optique). Les annexes de l’œil (glandes lacrymales, conjonctive) sont une localisation classique des lymphomes de type MALT.

Tableau 11-6 Classification d’Ann Arbor.
Les index pronostiques diffèrent selon l’histologie. Pour les LNH agressifs, l’index pronostique international (IPI) dépend du stade d’Ann Arbor, du PS, du taux de LDH (lacticodéshydrogénase) initial, de l’âge et du nombre d’atteintes extraganglionnaires. L’index pronostique international pour les lymphomes folliculaires (follicular lymphoma international prognostic index ou FLIPI) intègre le stade, le taux de LDH, le taux d’hémoglobine, le nombre de sites ganglionnaires atteints et l’âge (tableau 11-7) [9].

Tableau 1-7 Lymphomes malins non hodgkiniens, index pronostique international (IPI).
LDH : lacticodéshydrogénase ; PS : performance status (état général).
Avec les traitements plus récents associant le rituximab à la chimiothérapie, l’IPI garde sa valeur pronostique mais semble n’identifier que trois groupes :
un groupe avec zéro facteur de très bon pronostic (survie globale à 4 ans de 94 %) ;
un groupe avec un ou deux facteurs de bon pronostic (survie globale à 4 ans de 79 %) ;
un groupe avec trois à cinq facteurs de moins bon pronostic (survie globale à 4 ans de 55 %) [11].

Tableau 11-8 Survie en fonction de l’IPI tous âges confondus.
Le traitement repose sur la polychimiothérapie séquentielle associée aux anticorps monoclonaux anti-CD20 dans les lymphomes de type B. Le type de polychimiothérapie dépend du type histologique et de l’âge. La polychimiothérapie classique de type CHOP (cyclophosphamide, doxorubicine, vincristine et prednisone) des LNH diffus à grandes cellules B associe une anthracycline, du cyclophosphamide, de l’oncovin et des corticoïdes. La chirurgie d’exérèse n’a pas de place dans le traitement des lymphomes. La place de la radiothérapie est controversée selon les études dans les LNH agressifs. Un traitement de consolidation par chimiothérapie intensive avec support hématopoïétique peut être indiqué dans certaines situations.
Schématiquement, les traitements actuels des LNH diffus à grandes cellules B chez les patients de moins de 60 ans, et selon l’IPI, ont un taux de réponse complète compris entre 60 et 90 %, et une survie long terme comprise entre 55 et 90 %.
Des progrès majeurs ont été accomplis ces dernières années et démontrés grâce à des études prospectives multicentriques :
importance de l’association de l’anti-CD20 à la chimiothérapie dans les LNH diffus à grandes cellules B [12] ;
intérêt du traitement d’entretien par anti-CD20 pour les LNH folliculaires en bonne réponse thérapeutique après le traitement d’induction [13] ;
intérêt de l’aracytine à haute dose et d’une consolidation par intensification thérapeutique avec support hématopoïétique pour les LNH du manteau [14] ;
intérêt d’un traitement, selon les schémas pédiatriques, pour les LNH de type Burkitt de l’adulte [15].
Les études en cours concernant par exemple les LNH diffus à grandes cellules B tendent à une désescalade ou à une intensification des traitements selon la rapidité et la qualité de la réponse thérapeutique évaluée par la réponse métabolique mesurée par le PET scan au fluor FDG (fluorodésoxyglucose).
Les localisations intra-oculaires des lymphomes malins peuvent également être liées à des localisations secondaires de lymphomes ganglionnaires de haut grade de malignité. Lorsqu’il s’agit d’une localisation secondaire, les cellules lymphomateuses atteignent l’œil par la circulation choroïdienne. L’atteinte choroïdienne est donc prédominante avec une infiltration de la choriocapillaire, une ischémie de l’épithélium pigmenté et un décollement exsudatif rétinien. On peut également avoir une infiltration de l’uvée antérieure avec une infiltration de l’iris et des « uvéites antérieures » cortico-résistantes [16]. Le plus souvent, le lymphome ganglionnaire est déjà connu, le diagnostic est donc aisé. L’atteinte oculaire inaugurale d’un lymphome malin systémique est exceptionnelle. Le diagnostic différentiel peut être la maladie de Vogt-Koyanagi-Harada, la sclérite postérieure, voire une choroïdite séreuse centrale. Le bilan étiologique permet souvent de faire le diagnostic dès la numération formule sanguine qui objective souvent des cellules tumorales circulantes [17]. Certaines formes peuvent être particulièrement difficiles à diagnostiquer. L’atteinte rétinienne nécrosante, et hémorragique en particulier, peut être liée au lymphome. Ces patients étant immunodéprimés par les traitements du lymphome peuvent présenter également une toxoplasmose oculaire extensive ou une nécrose virale de la rétine à herpèsvirus ou cytomégalovirus (CMV). Le diagnostic correct est alors fondamental car en l’absence de traitement adéquat, la vision peut être compromise. Dans ces cas, une ponction de la chambre antérieure ou une vitrectomie diagnostique, avec recherche des virus et de la toxoplasmose par polymerase chain reaction (PCR), sont très importantes.
Les lymphomes de la zone marginale (marginal zone lymphoma ou MZL) représentent un groupe de lymphomes dont l’origine cellulaire dérive de lymphocytes B issus de la zone marginale, correspondant à une région anatomique spécifique des organes lymphoïdes secondaires. Ces cellules peuvent être issues d’une région anatomiquement localisée dans des organes lymphoïdes (rate et ganglions) ou dans des organes non lymphoïdes au niveau de muqueuses associées aux tissus lymphoïdes (MALT). Il existe enfin des atteintes extranodales de tissus non associés aux muqueuses telles que la peau, l’orbite ou la dure-mère.
Le groupe international d’études des lymphomes extranodaux (International Extranodal Lymphoma Study Group ou IELSG) distingue trois sous-groupes de MZL en fonction de l’atteinte initiale de la lésion :
Le lymphome extranodale MZL de type MALT ;
le lymphome splénique de la zone marginale (avec ou sans lymphocyte villeux) ;
le lymphome de la zone marginale nodal (avec ou sans cellules monocytoïdes B) [18].
Les lymphomes de la zone marginale représentent entre 5 et 17 % de l’ensemble des lymphomes non hodgkinien (LNH) de l’adulte selon les séries.
Le lymphome de type MALT est le plus fréquent des sous-types de lymphomes de la zone marginale, représentant entre 50 et 70 % des MZL soit 8 % environ des LNH, le plaçant au troisième rang des LNH de l’adulte. La médiane d’âge est de 60 ans au diagnostic, avec une prépondérance modérée chez la femme. Certaines études ont montré une variabilité de l’incidence en fonction de zones géographiques, par exemple on note une incidence augmentée dans le nord de l’Italie du lymphome de MALT de localisation gastrique. Il est également intéressant de noter que l’incidence de la localisation gastrique est en baisse alors qu’elle augmente régulièrement dans les autres localisations dont l’œil avec des sujets plus jeunes [19].
Ce sous-type anatomo-pathologique de lymphome de type MZL diffère des autres sous-types (splenic marginal zone lymphoma, nodal marginal zone lymphoma), en se développant dans des organes où il n’existe pas physiologiquement de tissu lymphoïde.
Six agents bactériens sont maintenant identifiés comme agents pathogènes associés aux lymphomes de la zone marginale de type MALT. Helicobacter pylori est le mieux caractérisé d’entre eux, associé au lymphome gastrique de type MALT. Pour les formes extradigestives oculaires, le lien entre l’infection à Chlamydophila psittaci et l’atteinte lymphomateuse orbitaire est très discuté. La bactérie n’a été retrouvée associée à la localisation conjonctivale que dans quelques régions du globe [19].
Cette stimulation antigénique chronique, par des auto-antigènes et/ou des agents pathogènes microbiens, induit une prolifération polyclonale B initiale et permet d’expliquer l’apparition d’infiltrats lymphoïdes dans des sites extranodaux [20].
Cette stimulation immunitaire induit une accumulation de tissus lymphoïdes dans des sites typiques pour chaque entité de lymphomes des muqueuses.
Les lymphomes de MALT présentent des aberrations chromosomiques récurrentes, dont des translocations chromosomiques, ou des aberrations équilibrées.
La translocation t(11;18)(q21;q21) est la plus fréquente des anomalies chromosomiques structurales dans le lymphome de MALT ; présente dans 10 à 50 % des lymphomes gastriques de type MALT, elle est rarement retrouvée dans les autres formes de lymphome non hodgkinien, à l’exception de l’atteinte pulmonaire.
La présence de cette translocation t(11;18) (q21;q21) dans les MALT est corrélée avec la perte d’une instabilité génétique ou des déséquilibres chromosomiques.
L’accumulation d’anomalies génétiques permet une autonomisation de prolifération indépendamment de la stimulation antigénique chronique. Ces anomalies acquises peuvent induire la transformation de lymphomes à petites cellules en lymphomes à grandes cellules.
La présentation clinique varie au diagnostic selon la localisation de l’atteinte lymphomateuse, même si certaines caractéristiques sont habituellement retrouvées. La plupart des patients présentent au diagnostic :
un état général conservé ;
l’absence de signe clinique d’évolutivité : absence de sueurs nocturnes profuses, absence de perte de poids supérieure à 10 % du poids habituel en 6 mois et fébricule à prédominance vespérale sans étiologie infectieuse retrouvée depuis plus de 3 semaines ;
l’absence d’anomalie biologique de mauvais pronostic, telle l’élévation de la β2-microglobulinémie et du taux de LDH.
La maladie est localisée dans la majorité des cas (atteinte multifocale dans 30 à 40 % des cas).
La dissémination de la maladie peut se faire à d’autre atteinte muqueuse ou bien à partir d’un site muqueux vers une atteinte viscérale splénique, médullaire ou hépatique. L’atteinte médullaire est retrouvée chez 20 % des patients au diagnostic. Enfin le risque de dissémination est significativement augmenté chez les patients ayant une atteinte non digestive (thyroïde, poumon, glandes salivaires, peau, orbite).
Le staging des lymphomes indolents de type MALT quelle que soit la localisation peut se faire selon la classification de Ann Harbor utilisée pour les lymphomes d’une manière générale. Elle s’applique cependant mal aux lymphomes des annexes de l’œil et des orbites où une classification TNM (tumor-nodes-metastases) est plus appropriée [21]. La classification TNM (tableau 11-9) est établie après un bilan fait par les hématologistes.

Tableau 11-9 Classification TNM des lymphomes.
a L’orbite antérieur est défini comme la région située entre le septum orbitaire et l’équateur du globe oculaire. L’orbite postérieur est défini comme la région postérieure à l’équateur, allant jusqu’à l’apex orbitaire.
b Un envahissement palpébral est présent lorsque le lymphome des annexes oculaires infiltre les tissus préseptaux (c’est-à-dire les tissus antérieurs au septum orbitaire).
c Les nœuds lymphatiques régionaux, dont les nœuds pré-auriculaires (parotide), submandibulaires et cervicaux.
(Source : American Joint Committee on Cancer [AJCC], Chicago, Illinois.)
L’atteinte oculaire des lymphomes de bas grade (lymphome de MALT et plus rarement lymphome du manteau) est la plus fréquente des atteintes oculaires liées à un lymphome. Les données épidémiologiques montrent une augmentation d’incidence de ces lymphomes des annexes de l’œil de 6,3 % par an [19]. C’est la tumeur la plus fréquente des annexes oculaires représentant 55 % de celles-ci [22]. Les atteintes des annexes oculaires par le lymphome sont dues, par ordre décroissant, à des lymphomes extraganglionnaires de la zone marginale associée aux muqueuses (MALT), à des lymphomes folliculaires (environ 20 %) et à des lymphomes diffus à grandes cellules (environ 8 %). Plus rarement, on peut rencontrer des lymphomes du manteau, lymphomes lymphocytiques, lymphomes lymphoplasmocytaires. Enfin, 10 à 32 % des atteintes sont une atteinte secondaire à un lymphome systémique [23].
Ces localisations des annexes sont de gravité variables allant de la simple hyperplasie lymphocytaire bénigne réactionnelle au lymphome malin agressif.
Le diagnostic repose sur la biopsie d’une lésion qui révèle une prolifération dont l’organisation est caractéristique de petits lymphocytes B CD20+, CD10–, CD23–, BCL-6–, CD3+ et en général CD5–. La monoclonalité peut être retrouvée par les techniques de PCR et l’immuno-histochimie signale une translocation t(11;14)(q13;q32). D’autres translocations peuvent être découvertes, par exemple t(3;14)(p14;q32) retrouvée dans 20 % des cas qui juxtapose le gène FOXP1 au gène promoteur des IgH.
La physiopathogénie a fait l’objet de nombreux débats pour retrouver un agent pathogène déclenchant la prolifération lymphocytaire. Si Helicobacter pilori a été démontré comme acteur dans le lymphome de l’estomac, cela reste non établi dans le lymphome des annexes. Certaines équipes ont montré une association entre le MALT de la conjonctive et l’ADN de Chlamydophila psittaci (la plupart du temps en Asie) mais d’autres équipes ne retrouvent pas ces résultats, en particulier en Europe [24]. Il n’est pas actuellement démontré que le MALT de la conjonctive est associé à une infection bactérienne quelle qu’elle soit [25].
Sur le plan clinique, les sites atteints sont de préférence : l’orbite (40 %) induisant un syndrome orbitaire suivi de la conjonctive (35-40 %) produisant une lésion conjonctivale souvent localisée dans les culs-de-sac rose saumoné assez typique ; la glande lacrymale (10-15 %) ; les paupières (10 %). En général, l’atteinte est unilatérale mais peu gagner les deux yeux simultanément ou de manière différée dans 10 à 15 % des cas [22, 23].
Les lésions sont d’évolution lente et le diagnostic peut être établi plusieurs années après le début des symptômes, en particulier dans la forme conjonctivale.
Le diagnostic repose sur la biopsie des lésions qui ne doivent pas être fixées mais données à l’état frais en anatomo-pathologie pour pouvoir réaliser les immuno-marquages.
À côté de ces atteintes des annexes de l’œil ou de l’orbite, il existe une atteinte beaucoup plus rare correspondant à une infiltration de l’uvée. L’atteinte de l’uvée antérieure est exceptionnelle et induit une infiltration de l’iris ou du corps ciliaire ; l’atteinte de l’uvée postérieure se présente comme une infiltration de la choroïde jaunâtre, la choroïde est épaissie de façon plus ou moins diffuse avec des plis choroïdiens et un décollement exsudatif rétinien. L’échographie oculaire B montre un épaississement choroïdien plutôt diffus avec parfois un épaississement en arrière de la sclère. Le diagnostic à ce stade est souvent erroné, on évoque une sclérite postérieure, une pseudo-tumeur inflammatoire de l’orbite ou une métastase choroïdienne. Il est aussi souvent fait tardivement après échec d’un traitement corticoïde ou immunosuppresseur pour une supposée sclérite et un bilan d’extension négatif excluant un carcinome primitif. Le diagnostic repose sur la mise en évidence des cellules tumorales soit ab externo par une biopsie de la sclère soit ab interno par une biopsie choroïdienne (fig. 11-29) [26].
Une fois le diagnostic établi, un bilan d’extension s’impose qui sera fait par les hématologues. Il existe également une classification TNM pour l’atteinte oculaire des lymphomes de MALT [21]. Celle-ci semble plus appropriée que la classique classification de Ann Arbor pour cette localisation (voir plus haut).

Fig. 11-28 Atteinte conjonctivale d’un lymphome de type MALT.

Fig. 11-29 Atteinte choroïdienne par un lymphome de MALT de bas grade.
En dépit d’une abondante littérature sur les caractéristiques physiopathologiques des lymphomes de MALT, il n’existe que quelques séries rétrospectives où il est question de la place de la chirurgie, de la chimiothérapie ou de la radiothérapie. L’IELSG a permis de coordonner cependant quelques études prospectives.
L’oncogenèse unique de ce type de maladie, liée à des co-facteurs pathogènes impliqués dans l’initiation de l’hémopathie, a un impact sur le traitement. Pour les maladies localisées, l’éradication d’Helicobacter pylori au niveau gastrique par une antibiothérapie ciblée permet l’obtention d’une régression complète lymphomateuse dans environ 80 % des cas.
Dans les localisations extragastriques comme la conjonctive, le traitement local antibiotique n’a jamais démontré d’intérêt.
Dans les formes localisées, une radiothérapie ou une chirurgie peuvent être indiquées, permettant un excellent contrôle de la maladie [27]. Pour les formes disséminées au diagnostic, l’utilisation de chimiothérapies de type alkylant (Chloraminophène® ou Alkéran®) ou des analogues des purines (fludarabine) permet d’induire des taux de réponse complète dans 75 % des cas, avec une EFS (event free survival) et une OS (overall survival) à 5 ans respectivement à 50 % et 75 %. Les anthracyclines ne sont classiquement utilisées qu’en cas de transformation du lymphome indolent en lymphome agressif à grandes cellules B ou en cas de critères de forte masse tumorale définie par une masse de taille supérieure à 7 cm et/ou un taux élevé de LDH.
L’activité clinique de l’anticorps monoclonal anti-CD20 de type MabThera® a été démontrée dès 2003, dans les atteintes nodales et extranodales de lymphomes de la zone marginale, avec une réponse tumorale de l’ordre de 75 % lorsqu’il est utilisé en première ligne thérapeutique. Les patients atteints de lymphome de type MALT ont une évolution favorable avec une survie à 5 ans entre 86 et 95 % sans différence significative entre les formes digestives et les formes non gastro-intestinales d’une part et entre les formes localisées et les formes disséminées d’autre part.
Le traitement de la forme oculaire isolée ne repose sur aucun consensus. En fonction des équipes, il est basé sur la radiothérapie seule, l’association à une chimiothérapie ou l’immunothérapie par les anticorps anti-CD20 qui est à discuter en fonction du bilan et avec l’hématologiste. Certaines équipes préconisent également un traitement antibiotique anti-Chlamydia. Aucune de ces stratégies n’a fait l’objet d’une étude randomisée efficacité versus toxicité et les protocoles thérapeutiques varient en fonction des équipes [22, 25, 27].
Il est de règle de ne faire de chirurgie d’exérèse que dans des formes très localisées, ce qui est rarement le cas. En faisant l’exérèse ou la biopsie de ces lésions conjonctivales, il faut faire deux prélèvements. L’un sera fixé, l’autre sera congelé pour l’immuno-histochimie. La plupart du temps, les lésions conjonctivales sont étendues prenant souvent tout le cul-de-sac conjonctival. Dans ce cas, il ne faut pas faire une exérèse délabrante mais une biopsie. Le choix thérapeutique dépend de la forme histologique et du staging hématologique et pourra être une immunothérapie par anticorps anti-CD20, une radiothérapie externe, une chimiothérapie ou une combinaison de ces différents traitements [28].
L’atteinte orbitaire ou plus exceptionnellement conjonctivale des adénocarcinomes métastatiques reste un événement rare. Il s’agit en général de patients atteint d’un adénocarcinome (sein, poumon) ou d’un carcinome neuro-endocrine dans le cadre de l’évolution métastatique de leur maladie. De même les leucémies en rechute peuvent présenter des infiltrations conjonctivales ressemblant à une infiltration lymphomateuse. La maladie étant connue, le diagnostic n’est pas difficile et peut être confirmé par une seule biopsie. Les infiltrations conjonctivales métastatiques sont généralement des infiltrations achromes plus ou moins hémorragiques sauf dans le cas très rare de métastases sous-conjonctivales d’un mélanome malin cutané métastatique où la lésion est fortement pigmentée (fig. 11-30).
Le traitement repose sur le traitement de la maladie initiale et sur une éventuelle radiothérapie adjuvante.
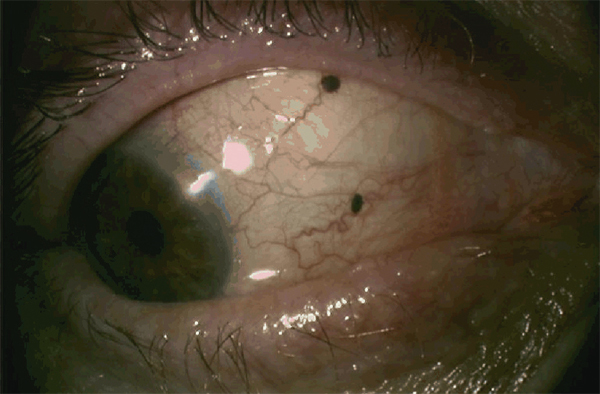
Fig. 11-30 Métastases conjonctivales d’un mélanome cutané métastatique.
[1] Coupland SE, Hummel M, Stein H. Ocular adnexal lymphomas : five case presentations and a review of the literature. Surv Ophthalmol 2002 : 47 : 470-90.
[2] Mateo-Montoya A, et al. White dots in the eye fundus revealing Hodgkin’s lymphoma. Eye 2009 ; 24 : 934-7.
[3] Thakker MM et al. Multifocal nodular episcleritis and scleritis with undiagnosed Hodgkin’s lymphoma. Ophthalmology 2003 ; 110 : 1057-60.
[4] Campo E, et al. The 2008 WHO classification of lymphoid neoplasms and beyond : evolving concepts and practical applications. Blood 2011 ; 117 : 5019-32.
[5] Cultrera JL, Dalia SM. Diffuse large B-cell lymphoma : current strategies and future directions. Cancer 2011 ; 19 : 204-13.
[6] Gromminger S, Mautner J, Bornkamm GW. Burkitt lymphoma : the role of Epstein-Barr virus revisited. Br J Haematol 2012 ; 156 : 719-29.
[7] Arcaini L, et al. Indolent B-cell lymphomas associated with HCV infection : clinical and virological features and role of antiviral therapy. Clin Dev Immunol 2012 ; 2012 : 638185.
[8] Conteduca V, et al. H. pylori infection and gastric cancer : state of the art (review). Int J Oncol 2013 ; 42 : 5-18.
[9] Solal-Celigny P et al. Follicular lymphoma international prognostic index. Blood 2004 ; 104 : 1258-65.
[10] A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. N Engl J Med 1993 ; 329 : 987-94.
[11] Sehn LH, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood 2007 ; 109 : 1857-61.
[12] Coiffier B, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients : a study by the Groupe d’Études des Lymphomes de l’Adulte. Blood 2010 ; 116 : 2040-5.
[13] Salles G, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA) : a phase 3, randomised controlled trial. Lancet 2011 ; 377(9759) : 42-51.
[14] Delarue R, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation (ASCT) in mantle cell lymphoma (MCL) : a phase 2 study from the GELA. Blood 2013 ; 121 : 48-53.
[15] Divine M, et al. Burkitt lymphoma in adults : a prospective study of 72 patients treated with an adapted pediatric LMB protocol. Ann Oncol 2005 ; 16 : 1928-35.
[16] Mashayekhi A, Shields CL, Shields JA. Iris involvement by lymphoma : a review of 13 cases. Clin Experiment Ophthalmol 2013 ; 41 : 19-26.
[17] Sjo LD. Ophthalmic lymphoma: epidemiology and pathogenesis. Acta Ophthalmol 2009 ; 87 : 1-20.
[18] Zucca E, et al. Marginal zone lymphomas. Hematol Oncol Clin North Am 2008 ; 22 : 883-901, viii.
[19] Khalil MO, et al. Incidence of marginal zone lymphoma in the United States, 2001-2009 with a focus on primary anatomic site. Br J Haematol 2014 ; 165 : 67-77.
[20] Peveling-Oberhag J, et al. Hepatitis C-associated B-cell non-Hodgkin lymphomas. Epidemiology, molecular signature and clinical management. J Hepatol 2013 ; 59 : 169-77.
[21] Coupland SE, et al. A TNM-based clinical staging system of ocular adnexal lymphomas. Arch Pathol Lab Med 2009 ; 133 : 1262-7.
[22] Rasmussen PK, et al. Ocular adnexal follicular lymphoma : a multicenter international study. JAMA Ophthalmol 2014 ; 132 : 851-8.
[23] Charlotte F, et al. Ocular adnexal marginal zone B cell lymphoma : a clinical and pathologic study of 23 cases. Virchows Arch 2006 ; 448 : 506-16.
[24] Matthews JM, et al. Ocular adnexal Lymphoma : no evidence for bacterial DNA associated with lymphoma pathogenesis. Br J Haematol 2008 ; 142 : 246-9.
[25] Decaudin D, et al. Ocular adnexal lymphoma and Helicobacter pylori gastric infection. Am J Hematol 2010 ; 85 : 645-9.
[26] Loriaut P, et al. Choroidal and adnexal extranodal marginal zone B-cell lymphoma : presentation, imaging findings, and therapeutic management in a series of nine cases. Eye (Lond) 2013 ; 27 : 828-35.
[27] Tran KH, et al. Efficacy of low dose radiotherapy for primary orbital marginal zone lymphoma. Leuk Lymphoma 2012 ; 54 : 491-6.
[28] Zucca E, et al. Addition of rituximab to chlorambucil produces superior event-free survival in the treatment of patients with extranodal marginal-zone B-cell lymphoma : 5-year analysis of the IELSG-19 Randomized Study. J Clin Oncol 2013 ; 31 : 565-72.
C. Levy-Gabriel
La caroncule est composée d’un épithélium non kératinisé similaire à celui de la conjonctive, d’éléments cutanés (follicules pileux, glandes sébacées et sudoripares) et de glandes lacrymales accessoires. Toutes les lésions tumorales pouvant survenir au niveau de la peau, de la conjonctive ou des glandes lacrymales pourront donc apparaître au niveau de la caroncule [1, 2]. Dans cette localisation, 95 % des lésions sont bénignes, les plus fréquemment rencontrées étant le nævus (45 %) et le papillome (15 %). Les autres lésions caronculaires classiques sont le granulome pyogénique, le kyste d’inclusion, l’hyperplasie sébacée, l’adénome sébacé et l’oncocytome. Les tumeurs malignes comme le carcinome épidermoïde, le mélanome, le lymphome et le carcinome sébacé sont rares, représentant moins de 5 % des lésions caronculaires.
Les tumeurs épithéliales, mélanocytaires, lymphomateuses sont traitées dans les autres sous-chapitres de ce chapitre 11. Nous ne détaillerons donc ici que les tumeurs glandulaires propres à la caroncule qui se développent à partir :
des glandes lacrymales accessoires (oncocytome) ;
des glandes sébacées (hyperplasie sébacée, adénome sébacé et carcinome sébacé) ;
des glandes sudoripares (dacryoadénome).
L’oncocytome est une prolifération bénigne de l’épithélium des glandes lacrymales accessoires et apocrines, c’est donc un adénome. Il apparaît typiquement au niveau de la caroncule, mais peut aussi occasionnellement apparaître ailleurs au niveau de la conjonctive. C’est une tumeur peu fréquente, ne représentant que 4 % des tumeurs de la caroncule [3]. Le plus souvent, l’oncocytome survient chez une femme âgée. Cliniquement, il se présente comme un nodule vascularisé, parfois kystique, brun à rouge (fig. 11-31a) qui peut poser des problèmes de diagnostic différentiel avec une lésion épidermoïde, mélanocytaire, lymphocytaire ou amyloïde. La lésion est asymptomatique et grossit lentement [4]. En histologie, elle est composée d’une prolifération de cellules épithéliales, similaires en apparence à celles d’un épithélium apocrine (glandes de Moll), entourant un espace comme dans une glande (fig. 11-31b). Du fait de l’aspect kystique de ces espaces, le terme cystadénome apocrine est aussi utilisé pour décrire cette lésion. Les cellules épithéliales présentant un cytoplasme éosinophile, la lésion est aussi référencée comme un cystadénome éosinophile.
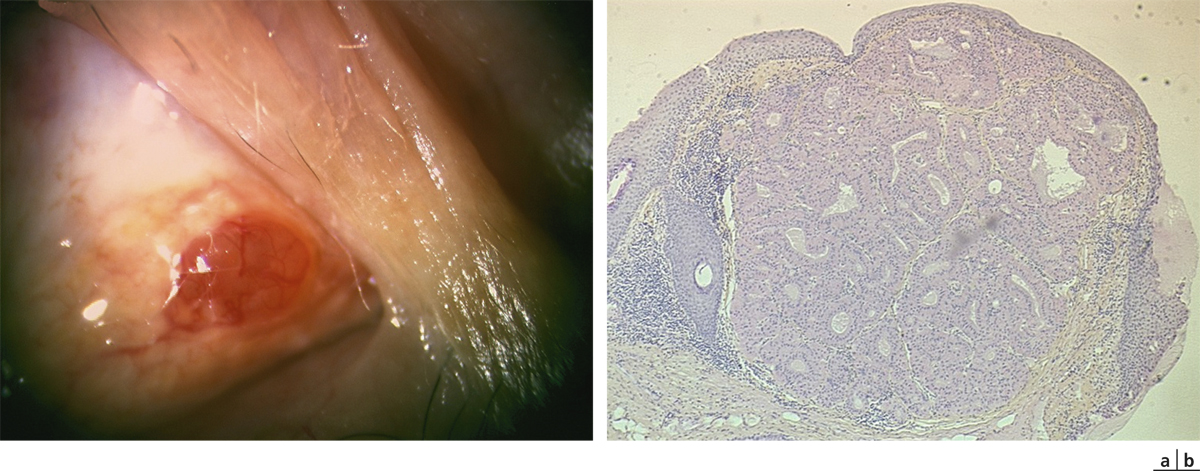
Ces deux lésions sont bénignes et rares. Elles ont une présentation clinique similaire avec un aspect de tuméfaction caronculaire jaune souple, parfois multinodulaire (fig. 11-32a). En histologie, l’aspect de l’hyperplasie sébacée est celui d’une glande sébacée unique mais hyperplasique avec de nombreux lobules glandulaires s’abouchant dans un canal central unique. L’adénome sébacé, lui, est composé de multiples lobules sébacés irréguliers et incomplètement différenciés (fig. 11-32b). Lorsqu’un adénome sébacé est diagnostiqué, un syndrome de Muir-Torre doit être recherché (affection autosomale dominante caractérisée par la survenue d’adénome ou d’adénocarcinome des glandes sébacées, de kératoacanthome ou de tumeurs gastro-intestinales).
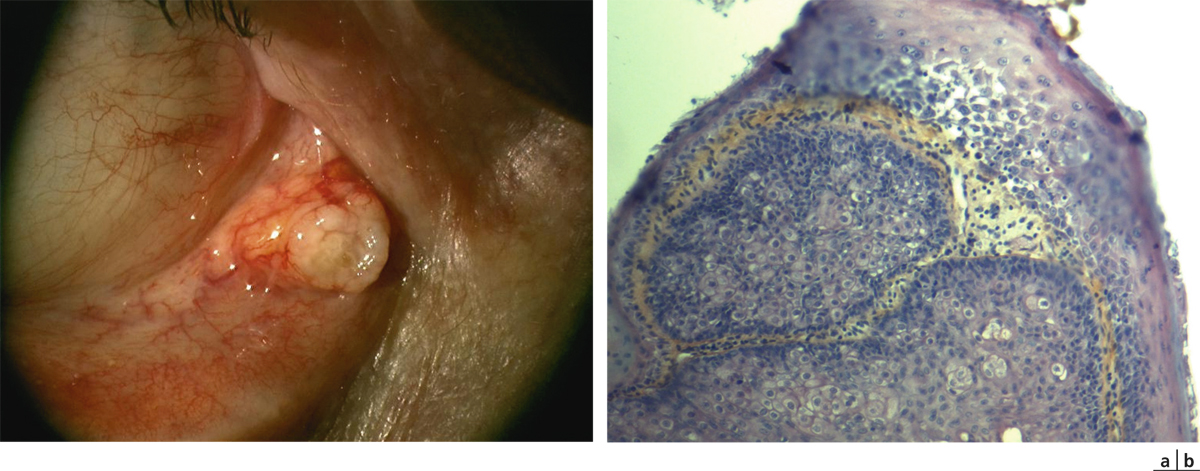
Le carcinome sébacé est une tumeur maligne développée à partir des glandes sébacées, à haut risque métastatique. Il apparaît en général chez une personne âgée au niveau de la paupière supérieure, mais il peut aussi se développer à partir des glandes sébacées de la caroncule. Il existe deux formes : la forme nodulaire palpébrale ou caronculaire (fig. 11-33a) et la forme évoluant en nappes au niveau de la conjonctive correspondant à un envahissement tumoral de l’épithélium conjonctival et parfois de la cornée sous une forme pagétoïde (fig. 11-34) [6]. Cette forme conjonctivale, avec croissance pagétoïde en nappes parfois multicentriques, se présente comme une lésion diffuse d’aspect inflammatoire à développement particulièrement insidieux. L’aspect clinique initial du carcinome sébacé prête donc souvent à confusion, posant des problèmes de diagnostic différentiel avec un chalazion pour la forme nodulaire palpébrale ou avec une blépharoconjonctivite chronique pour les formes évoluant en nappes au niveau de la conjonctive. Dans la série de Shields, 20 % des patients se présentent avec un diagnostic de chalazion et 25 % avec un diagnostic de blépharoconjonctivite [7, 8]. Il est donc recommandé de pratiquer une biopsie avec examen histologique devant tout chalazion récidivant ou toute blépharoconjonctivite unilatérale résistant au traitement médical.
En histologie, les carcinomes sébacés bien différenciés sont aisément identifiables par l’aspect spumeux et microvésiculaire du cytoplasme des cellules tumorales (fig. 11-33b). Les tumeurs peu différenciées sont parfois de diagnostic plus difficile et pourront être confondues avec les autres tumeurs épithéliales malignes plus fréquentes. Un aspect caractéristique mais non pathognomonique du carcinome sébacé est la dissémination de cellules tumorales individuelles ou en groupe au niveau de l’épithélium conjonctival, appelée infiltration pagétoïde. Cette infiltration pagétoïde peut dans certains cas entraîner un remplacement complet de l’épithélium conjonctival par des cellules tumorales (carcinome sébacé in situ).
Le traitement est avant tout chirurgical avec exérèse tumorale complète. En cas d’atteinte palpébrale, l’exérèse consistera en une résection palpébrale transfixiante ; des marges latérales de 5 mm de tissus sains sont conseillées. En cas d’aspect suspect d’envahissement conjonctival diffus pagetoïde, de multiples biopsies seront réalisées pour définir précisément l’étendue des lésions et adapter le traitement. Shields insiste sur l’importance des traitements combinés associant la cryothérapie, les chimiothérapies topiques ou la radiothérapie à la chirurgie initiale, en particulier en cas de lésions avancées [8]. En cas de récidive de lésions diffuses ou d’envahissement orbitaire, une exentération orbitaire pourra être nécessaire. Un diagnostic précoce est donc essentiel. Le pronostic vital des carcinomes sébacés est moins bon que celui des carcinomes épidermoïdes, avec une mortalité à 5 ans proche de 25 % [9]. Les facteurs de mauvais pronostic sont : un envahissement vasculaire, lymphatique ou orbitaire ; une faible différenciation ; une tumeur de plus de 10 mm ; une importante composante invasive ou un envahissement pagetoïde.

Fig. 11-33 Carcinome sébacé caronculaire.
Aspects en lampe à fente (a) et histologique avec aspect spumeux des cellules tumorales (b).

Fig. 11-34 Carcinome sébacé avec envahissement pagétoïde de la conjonctive du cul-de-sac inférieur.
Cette tumeur conjonctivale bénigne rare de l’enfant et l’adulte jeune a été décrite par Jakobiec [10]. Cliniquement, le dacryoadénome se présente au niveau de la conjonctive bulbaire ou palpébrale sous la forme d’une tuméfaction rose et charnue difficile à différencier d’un lymphome. En histologie, la tumeur semble prendre son origine au niveau de l’épithélium de surface, prolifère en profondeur dans le stroma et développe des lobules glandulaires analogues à ceux d’une glande lacrymale normale à part la présence de nombreuses cellules caliciformes. Le diagnostic clinique étant difficile, une exérèse chirurgicale est préférable. Le pronostic est excellent.
[1] Luthra CL, Doxanas MT, Green WR. Lesions of the caruncle : a clinicohistopathologic study. Surv Ophthalmol 1978 ; 23 : 183-95.
[2] Kaeser PF, et al. Tumors of the caruncle : a clinicopathologic correlation. Am J Ophthalmol 2006 ; 142 : 448-55.
[3] Shields CL, et al. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004 ; 111 : 1747-54.
[4] Carbonaro F, et al. Three oncocytomas in a short space of time. Br J Ophthalmol 2007 ; 91 : 1718-9.
[5] Sakuma T, Ueno C, Kawano K. Sebaceous adenoma of caruncula lacrimalis : report of two cases. Klin Monbl Augenheilkd 2009 ; 226 : 432-3.
[6] Pandey K, et al. Primary sebaceous gland carcinoma of the bulbar conjunctiva without involvement of eyelid : a clinical dilemma. Oman J Ophthalmol 2011 ; 4 : 97-9.
[7] Shields JA, et al. Sebaceous carcinoma of the eyelids : personal experience with 60 cases. Ophthalmology 2004 ; 111 : 2151-7.
[8] Shields JA, et al. Sebaceous carcinoma of the ocular region : a review. Surv Ophthalmol 2005 ; 50 : 103-22.
[9] Rao NA, et al. Sebaceous carcinomas of the ocular adnexa : a clinicopathologic study of 104 cases, with five-year follow-up data. Hum Pathol 1982 ; 13 : 113-22.
[10] Jakobiec FA, et al. Dacryoadenoma. A unique tumor of the conjunctival epithelium. Ophthalmology 1989 ; 96 : 1014-20.
C. Levy-Gabriel
Les tumeurs vasculaires représentent 4 % des tumeurs de la conjonctive [1].
Le granulome pyogénique est une lésion relativement commune, représentant 1 % de l’ensemble des tumeurs de la conjonctive [2] et 20 % des lésions vasculaires [3].
La terminologie classique de granulome pyogénique est en fait inexacte car la lésion n’est ni nécrotique, ni un véritable granulome. Il s’agit plutôt d’un tissu de granulation exubérant, c’est-à-dire une masse pédiculée composée d’un mélange de cellules inflammatoires aiguës et chroniques (lymphocytes, plasmocytes, neutrophiles), avec prolifération capillaire classiquement radiaire [4]. Certains auteurs préfèrent l’expression « hémangiome capillaire acquis de type granulome pyogénique » [5]. Cette prolifération fibrovasculaire apparaît en réponse à un traumatisme, chirurgical ou accidentel : contexte de chalazion (au niveau de la conjonctive palpébrale), antécédent de chirurgie du strabisme (au niveau de l’insertion musculaire), chirurgie rétinienne, énucléation, pose de clous lacrymaux, port de lentilles de contact [6, 7]. Il apparaît cliniquement comme un nodule en relief, rouge, d’apparition récente et de croissance rapide, situé en général au niveau de la conjonctive, plus rarement au niveau de la cornée. La lésion est souple voire flasque, de forme très variable selon les cas, souvent pédiculée, plus rarement sessile. Le granulome pyogénique régresse parfois sous traitement corticoïde topique, mais une exérèse chirurgicale est souvent nécessaire. Shields réalise une incision au niveau de la base associée à une cautérisation ou une cryo-application et rapporte de bons résultats. En cas de récidives multiples, une curiethérapie à faibles doses peut être efficace [8].
Les lymphangiectasies correspondent à la présence de vaisseaux lymphatiques dilatés et proéminents au niveau de la conjonctive (fig. 11-35). Si les lymphangiectasies prennent un aspect tumoral on parle alors de lymphangiome. Les lymphangiectasies et lymphangiomes sont des tumeurs conjonctivales rares correspondant à moins de 1 % des cas d’une série de 1643 tumeurs conjonctivales publiée par Shields [2].
Le lymphangiome représente 20 % des tumeurs vasculaires conjonctivales [3]. Il se présente sous la forme d’une masse conjonctivale rouge, molle, constituée de vaisseaux lymphatiques de taille variable, à contenu souvent hémorragique. L’atteinte conjonctivale isolée est peu fréquente, dans la plupart des cas il s’agit de la partie superficielle visible d’un lymphangiome orbitaire. Une atteinte des fosses nasales peut être associée et responsable d’une épistaxis concomitante de l’hémorragie conjonctivale [9]. L’existence d’un contexte de lymphangiomatose systémique héréditaire est rare.
Les lymphangiectasies représentent 19 % des tumeurs vasculaires conjonctivales [3]. Elles sont caractérisées par la présence de vaisseaux lymphatiques linéaires à contenu clair ou sanguin, sans formation de masse.
Dans la plupart des cas, les patients sont surveillés régulièrement avec traitement symptomatique des complications locales hémorragiques. Les petites lésions localisées peuvent facilement être réséquées chirurgicalement. L’exérèse des lésions plus étendues est souvent plus difficile et compliquée d’importants saignements. Quelques publications ont rapporté des résultats satisfaisants après traitement de lymphangiomes non opérables par laser au dioxide de carbone ou, plus récemment, par curiethérapie au strontium 90 [10, 11].

Fig. 11-35 Lymphangiectasies.
L’angiome capillaire infantile représente 14 % des tumeurs vasculaires conjonctivales dans la série de Shields [3]. Il apparaît rapidement après la naissance, grossit progressivement pendant 2 ans puis régresse lentement, tout comme les angiomes capillaires palpébraux ou orbitaires auxquels il est parfois associé. En histologie, il est caractérisé par des proliférations de cellules endothéliales lobulaires séparées par un fin septum fibreux. Les lésions involuées sont moins vasculaires et plus fibreuses. Il se présente comme une petite masse rouge plus ou moins étendue. La plupart sont de petite taille et asymptomatiques, ne nécessitant qu’une surveillance [12]. En cas de lésion plus étendue avec risque d’amblyopie, un traitement corticoïde oral ou intralésionnel peut accélérer la régression. Les bêta-bloquants oraux ou topiques, utilisés récemment pour traiter les angiomes capillaires orbitaux palpébraux infantiles, n’ont pas encore été utilisés pour les atteintes palpébrales [13].
L’angiome caverneux représente 2,1 % des tumeurs vasculaires de la conjonctive [3]. La lésion conjonctivale stromale multilobulaire, rouge bleuté, de dimensions variables peut apparaître à n’importe quel âge. L’aspect clinique est parfois difficile à distinguer de celui d’un lymphangiome. L’angiome est isolé en général, l’association à un syndrome de Sturge-Weber ou une angiomatose diffuse néonatale sont plus rares. Une surveillance peut être proposée ou une exérèse chirurgicale. Si la limite postérieure de la lésion n’est pas visualisable cliniquement, un bilan par IRM sera utile pour déterminer l’importance de son extension. En histologie, l’angiome caverneux est composé de veines congestives et dilatées séparées par du tissu conjonctif. Des muscles lisses peuvent être présents dans les parois vasculaires [14, 15].
Le glomangiome est une lésion vasculaire relativement commune originaire du glomus neurovasculaire (structure spécialisée dans la thermorégulation). La localisation conjonctivale ou orbitaire antérieure est rare. L’aspect clinique est proche du lymphangiome avec une masse rouge bleuté. En histologie, le glomangiome est constitué de vaisseaux vides de sang, limités par des cellules cubiques similaires à celles du glomus neurovasculaire. En immuno-histochimie, ces cellules ne fixent pas les marqueurs spécifiques des cellules endothéliales, mais fixent les marqueurs antigéniques spécifiques du muscle. Une surveillance peut être proposée ou une exérèse chirurgicale [16].
N. Cassoux
Le sarcome de Kaposi, décrit en 1872 par un dermatologue hongrois, est une maladie cutanéomuqueuse qui atteint principalement les personnes âgées de souche méditerranéenne. Dans sa forme méditerranéenne, la maladie est indolente et essentiellement cutanée. La forme africaine est plus agressive pouvant s’accompagner d’adénopathie et d’une atteinte viscérale. Cette forme grave est souvent mortelle. Dans les années 1980, avec l’explosion de l’infection par le VIH chez les jeunes homosexuels, le nombre de malades atteints d’une maladie de Kaposi a fortement progressé. Elle touchait à l’époque environ 30 % des sujets atteints du syndrome d’immunodéficience acquise (sida) à un stade d’immunodéficience avec atteintes extensives multiples, muqueuses, ganglionnaires et viscérales aboutissant rapidement au décès [17].
L’atteinte muqueuse touche toutes les muqueuses y compris la conjonctive. L’atteinte de la conjonctive et des paupières est retrouvée chez 10 à 20 % des malades et peut être révélatrice de l’infection par le VIH. Au début, une lésion plate et violacée apparaît sur le versant cutané palpébral ou le versant muqueux de la paupière ou de la conjonctive. L’atteinte conjonctivale peut être révélée par des hémorragies sous-conjonctivales à répétition (fig. 11-36). La lésion augmente de taille pour devenir un nodule solide violacé. Le volume du nodule au niveau de la paupière peut entraîner un défaut d’occlusion palpébrale (fig. 11-37). L’analyse histologique révèle différents stades selon la classification de Dugel [18, 19] :
type 1 : des lésions plates avec dilatations vasculaires, correspondant à des tubes à endothélium aplati sans mitoses, entourées de cellules inflammatoires ;
type 3 : une lésion tumorale hémorragique constituée de cellules endothéliales fusiformes avec noyau hyperchromatique et de nombreuses mitoses.
La physiopathologie de cette affection a fait l’objet de nombreuses études [20]. L’infection du sujet par un virus de la famille des gamma-Herpesviridae, le HHV-8, est nécessaire à la genèse de la maladie. Cependant le sarcome de Kaposi n’apparaît pas chez tous les sujets infectés. L’existence d’un déficit immunitaire est un des facteurs favorisant son apparition chez des receveurs d’organes transplantés. Chez les personnes infectées par le VIH, au déficit immunitaire s’ajoute un effet additionnel propre au VIH dans la genèse du sarcome de Kaposi. Dans ce cas, plus de la moitié des personnes infectées par les deux virus développent la maladie. La prise en charge thérapeutique du sarcome de Kaposi chez les personnes infectées par le VIH doit tenir compte de ces mécanismes et, dans un premier temps, s’attacher à traiter l’infection par le VIH [21].
Avant l’instauration des traitements antirétroviraux efficaces contre le VIH, le traitement consistait en une résection des lésions les plus petites et un traitement par chimiothérapie des lésions les plus grosses ou des atteintes viscérales [22]. Cette chimiothérapie était inefficace. Le seul traitement efficace est la restauration immunitaire des patients capable de faire totalement régresser les lésions. Depuis l’instauration des traitements anti-VIH efficaces, l’incidence du sarcome de Kaposi a massivement baissé dans les pays qui disposent de ces traitements [23].

Fig. 11-36 Sarcome de Kaposi de la conjonctive bulbaire.

Fig. 11-37 Sarcome de Kaposi nodulaire de la paupière.
[1] Shields CL, Shields JA. Tumors of the conjunctiva and cornea. Surv Ophthalmol 2004 ; 49 : 3-24.
[2] Shields CL, et al. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004 ; 111 : 1747-54.
[3] Shields JA, et al. Vascular tumors of the conjunctiva in 140 cases. Ophthalmology 2011 ; 118 : 1747-53.
[4] Desjardins L. Anatomie pathologique en ophtalmologie. Tumeurs intra-oculaires. Issy-les-Moulineaux : Elsevier Masson/SFO ; 2011-2012, p. 61.
[5] Garg R, et al. Acquired capillary hemangioma of the eyelid in a child. J Pediatr Ophthalmol Strabismus 2009 ; 46 : 118-9.
[6] Ababneh OH, Msallam MM. Bilateral simultaneous pyogenic granuloma after perforated punctal plug insertion. Ophthal Plast Reconstr Surg 2014 ; 30 : e113-5.
[7] Ferry AP. Pyogenic granulomas of the eye and ocular adnexa : a study of 100 cases. Trans Am Ophthalmol Soc 1989 ; 87 : 327-43, discussion 343-7.
[8] Gunduz K, et al. Plaque radiation therapy for recurrent conjunctival pyogenic granuloma. Arch Ophthalmol 1998 ; 116 : 538-9.
[9] Quezada AA, et al. Lymphangioma of the conjunctiva and nasal cavity in a child presenting with diffuse subconjunctival hemorrhage and nosebleeds. J Pediatr Ophthalmol Strabismus 2007 ; 44 : 180-2.
[10] Behrendt S, Bernsmeier H, Randzio G. Fractionated beta-irradiation of a conjunctival lymphangioma. Ophthalmologica 1991 ; 203 : 161-3.
[11] Spector JA, Zide BM. Carbon dioxide laser ablation for treatment of lymphangioma of the conjunctiva. Plast Reconstr Surg 2006 ; 117 : 609-12.
[12] Ozdal PC, et al. Capillary hemangioma of the caruncle. Can J Ophthalmol 2004 ; 39 : 560-2.
[13] Shields CL, Shields JA. Conjunctival tumors in children. Curr Opin Ophthalmol 2007 ; 18 : 351-60.
[14] Kiratli H, et al. Recurrent subconjunctival hemorrhage due to cavernous hemangioma of the conjunctiva. Can J Ophthalmol 2012 ; 47 : 318-20.
[15] Malik A, et al. Isolated cavernous hemangioma of conjunctiva. Ophthal Plast Reconstr Surg 2010 ; 26 : 385-6.
[16] Kim HJ, et al. Fibrous histiocytoma of the conjunctiva. Am J Ophthalmol 2006 ; 142 : 1036-43.
[17] Boue F, Lebbe C. Kaposi’s sarcoma. Bull Cancer 2003 ; 90 : 393-8.
[18] Cassoux N. Œil et virus. Rapport Société francaise d’ophtalmologie. Paris : Masson ; 2000.
[19] Dugel PU, et al. Ocular adnexal Kaposi’s sarcoma in acquired immunodeficiency syndrome. Am J Ophthalmol 1990 ; 110 : 500-3.
[20] Tappero JW, et al. Kaposi’s sarcoma. Epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol 1993 ; 28 : 371-95.
[21] Moore PS. The emergence of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8). N Engl J Med 2000 ; 343 : 1411-3.
[22] Dugel PU, et al. Treatment of ocular adnexal Kaposi’s sarcoma in acquired immune deficiency syndrome. Ophthalmology 1992 ; 99 : 1127-32.
[23] Blum L, et al. Complete remission of AIDS-related Kaposi’s sarcoma associated with undetectable human herpesvirus-8 sequences during anti-HIV protease therapy. Aids 1997 ; 11 : 1653-5.
C. Levy-Gabriel
Le neurofibrome est une tumeur lentement progressive qui se développe à partir des gaines des nerfs périphériques. La localisation conjonctivale est très rare (un seul cas dans la cohorte de 1643 tumeurs conjonctivales rapportées par Shields) [1]. Comme au niveau orbitaire, le neurofibrome peut apparaître de façon isolée, diffuse ou sous la forme d’un neurofibrome plexiforme, ces deux dernières formes pouvant entrer dans le cadre d’une neurofibromatose de type 1 (NF1 ou maladie de Recklinghausen) [2-4]. Le neurofibrome conjonctival se présente comme une masse ferme, jaunâtre ou grisâtre, localisée au niveau du stroma conjonctival, sessile, à limites floues. La forme plexiforme est souvent le prolongement d’une atteinte palpébrale ou orbitaire et se présente comme une masse mal définie, irrégulière, souvent comparée à un sac de vers. En histologie, le neurofibrome est composé de fibroblastes endoneuraux, de cellules de Schwann et d’axones disposés en cordes et rubans au sein d’un stroma myxoïde. Il peut parfois être difficile à différencier des autres tumeurs à cellules fusiformes, et les colorations spéciales des axones peuvent dans certains cas être utiles. Les neurofibromes isolés avec croissance progressive peuvent bénéficier d’une exérèse chirurgicale complète. Le type plexiforme par contre peut être très difficile à enlever entièrement et la chirurgie se limite souvent à une simple réduction du volume tumoral.
Le schwannome (ou neurinome) est une tumeur bénigne qui comme le neurofibrome se développe à partir des gaines des nerfs périphériques, mais il est composé uniquement de cellules de Schwann. L’apparition d’un schwannome au niveau de la conjonctive est beaucoup plus rare qu’au niveau orbitaire (à peine une dizaine de cas publiés dans la littérature). Il peut être isolé ou associé à une NF1. Le schwannome conjonctival se présente comme une masse discrètement jaunâtre ou rosée, localisée au niveau du stroma conjonctival ou au niveau de l’épisclère (fig. 11-38). Cette lésion qui croît très lentement peut présenter quelques vaisseaux nourriciers conjonctivaux ou épiscléraux. L’exérèse chirurgicale complète de la tumeur avec sa capsule est conseillée pour éviter les récidives locales [5-7].
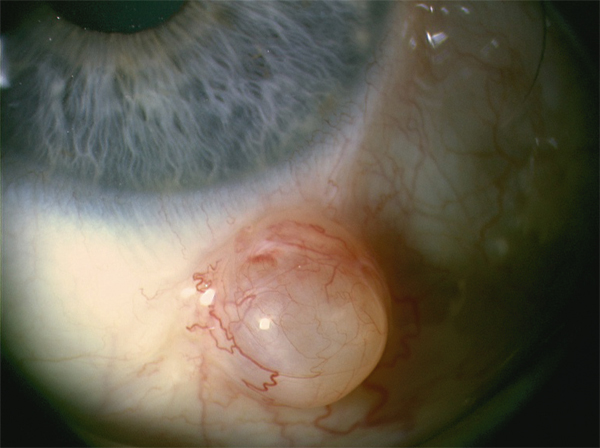
Fig. 11-38 Schwannome conjonctival.
Le fibrome conjonctival est une tumeur très rare (deux cas dans la série de 1643 tumeurs de Shields) composée de fibroblastes et de collagène. Il se présente sous la forme d’une masse blanche stromale, uni- ou multifocale [8, 9]. L’exérèse chirurgicale est conseillée.
L’histiocytome fibreux (autrefois appelé fibroxanthome) représente environ 1 % des tumeurs conjonctivales [1]. Il est composé de fibroblastes et d’histiocytes. Il peut être bénin, localement agressif ou malin [10-12]. En cas de malignité, la dissémination métastatique reste rare. Cliniquement, l’histiocytome fibreux se présente sous la forme d’une tumeur stromale ferme, rouge à brun, fréquemment localisée au limbe avec extension à la cornée (fig. 11-39). L’aspect est peu caractéristique et difficile à différencier des autres tumeurs stromales non pigmentées. L’exérèse chirurgicale complète de la lésion est conseillée.

Fig. 11-39 Histiocytome fibreux de la surface oculaire.
Bien qu’il puisse apparaître au niveau orbitaire, le lipome conjonctival est rare, en général il s’agit d’un lipome pléomorphe [13]. Il apparaît chez l’adulte, l’aspect est identique à celui d’un myxome (masse jaune : fig. 11-40). Il est composé d’un tissu conjonctif lâche et myxoïde et de lipocytes souvent fusiformes. Le traitement consiste en général en une exérèse chirurgicale.

Fig. 11-40 Lipome conjonctival.
Le xanthogranulome juvénile (XGJ) est une affection idiopathique rare liée à la prolifération non maligne de cellules histiocytaires non langerhansiennes. La manifestation clinique classique est une éruption cutanée de l’enfant avec papules transitoires jaunes à rouges, isolées ou multiples. Au niveau oculaire, les lésions iriennes sont les plus fréquemment décrites, souvent compliquées d’hyphéma ou de glaucome, mais le XGJ peut aussi affecter les paupières, les tissus orbitaires ou la conjonctive. L’atteinte conjonctivale est rare (22 cas publiés dans la littérature en 2009) [14]. Elle est en général isolée sans éruption cutanée associée et se présente sous la forme d’une masse en relief jaune souvent proche du limbe (fig. 11-41). Bien que le XGJ soit en général une affection de l’enfant, l’atteinte conjonctivale isolée survient souvent chez l’adulte. En histologie, le XGJ est composé d’histiocytes, de cellules inflammatoires chroniques et de cellules multinucléées (cellules géantes de Touton) avec un aspect typique d’anneau lipidique autour d’un foyer d’inflammation granulomateuse. Dans la plupart des cas, une exérèse chirurgicale est réalisée en raison du diagnostic clinique incertain [14, 15]. Néanmoins, si le diagnostic est suspecté cliniquement, des corticoïdes topiques ou généraux peuvent être proposés [16, 17].
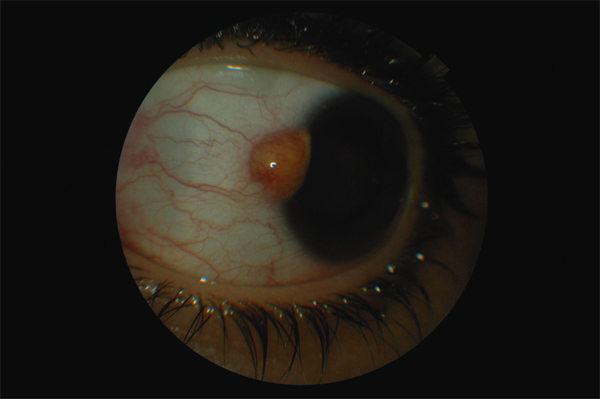
Le myxome est une tumeur bénigne d’origine mésenchymateuse. La localisation la plus classique est cardiaque. L’atteinte conjonctivale est rare, survient chez l’adulte, sous la forme d’une masse jaune ou rose, souple et mobile, en général située au niveau de la conjonctive bulbaire [18]. Dans la plupart des cas, les myxomes conjonctivaux sont isolés, mais ils peuvent parfois entrer dans le cadre d’un complexe de Carney, maladie autosomale dominante associant myxomes (cœur, peau), taches pigmentées de la peau et des muqueuses, suractivité endocrinienne et schwannomes. Face à un myxome conjonctival, une atteinte cardiaque doit donc systématiquement être recherchée. En anatomo-pathologie, le myxome est une lésion peu cellulaire avec des cellules stellaires et fusiformes disparates au sein d’un stroma myxoïde. Des vacuoles cytoplasmiques sont fréquemment retrouvées, quelques mastocytes peuvent être présents. Les colorations spéciales et la microscopie électronique seront parfois nécessaires pour différencier cette tumeur d’un lipome ou liposarcome, d’un neurofibrome myxoïde ou d’un rhabdomyosarcome. Le traitement consiste en général en une exérèse chirurgicale, mais les petites lésions asymptomatiques peuvent aussi être surveillées. Les récidives après exérèse chirurgicale sont rares.
[1] Shields CL, et al. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004 ; 111 : 1747-54.
[2] Freedman KA, Tran RM. Neurofibroma involving the caruncle. Arch Ophthalmol 2004 ; 122 : 294-5.
[3] Kalina PH, et al. Isolated neurofibromas of the conjunctiva. Am J Ophthalmol 1991 ; 111 : 694-8.
[4] Perry HD. Isolated neurofibromas of the conjunctiva. Am J Ophthalmol 1992 ; 113 : 112-3.
[5] Andreoli CM, et al. Perilimbal conjunctival schwannoma. Arch Ophthalmol 2004 ; 122 : 388-9.
[6] Charles NC, et al. Conjunctival neurilemoma. Report of 3 cases. Arch Ophthalmol 1997 ; 115 : 547-9.
[7] Demirci H, et al. Epibulbar schwannoma in a 17-year-old boy and review of the literature. Ophthal Plast Reconstr Surg 2010 ; 26 : 48-50.
[8] Charles NC, Ostriker GE. Epibulbar conjunctival fibroma. Ophthal Plast Reconstr Surg 2007 ; 23 : 249-50.
[9] Jakobiec FA, et al. Epibulbar fibroma of the conjunctival substantia propria. Arch Ophthalmol 1988 ; 106 : 661-4.
[10] Kim HJ, et al. Fibrous histiocytoma of the conjunctiva. Am J Ophthalmol 2006 ; 142 : 1036-43.
[11] Arora R, et al. Malignant fibrous histiocytoma of the conjunctiva. Clin Experiment Ophthalmol 2006 ; 34 : 275-8.
[12] Ryan G, Glasson B, Foster A. Benign fibrous histiocytoma of the conjunctiva. Case Rep Ophthalmol Med 2012 ; 2012 : 786260.
[13] Bryant J. Pleomorphic lipoma of the bulbar conjunctiva. Ann Ophthalmol 1987 ; 19 : 148-9.
[14] Soler V, et al. Juvenile xanthogranuloma of the corneoscleral limbus. J Fr Ophtalmol 2009 ; 32 : 436.e1-6.
[15] Mocan MC, et al. Juvenile xanthogranuloma of the corneal limbus: report of two cases and review of the literature. Cornea 2008 ; 27 : 739-42.
[16] Hermel M, Donner A, Remky A. New treatment option for adult-onset limbal xanthogranuloma. Cornea 2010 ; 29 : 113-6.
[17] De Keyser C, et al. Juvenile xanthogranuloma of the corneoscleral limbus : report of two cases. Ophthalmic 2011 ; 32 : 54-6.
[18] Demirci H, et al. Report of a conjunctival myxoma case and review of the literature. Arch Ophthalmol 2006 ; 124 : 735-8.
D. Brémond-Gignac
Les tumeurs congénitales du limbe sont les plus fréquentes des tumeurs de la surface oculaire de l’enfant ; elles sont constituées, dans la majeure partie, de dermoïdes du limbe. Ces derniers sont des choristomes épiscléraux [1]. Ils sont constitués d’une hypertrophie (ou de reliquats) de tissus normaux recouverts d’un épithélium épidermoïde avec une localisation anormale. Ceci survient habituellement lors de la fermeture des fissures fœtales, ce qui explique leur localisation toujours semblable. La localisation peut être limbique (dermoïdes du limbe) ou conjonctivale pure (dermolipomes). Ces lésions peuvent être uni- ou bilatérales et plus de 85 % sont situées au niveau de la conjonctive bulbaire, du limbe, de la cornée ou de la caroncule. Elles peuvent aussi être uniques ou multiples.
Les choristomes sont supposés se former chez l’embryon entre la 5e et 10e semaine de gestation, ils sont dus à une transformation métaplasique du mésoblaste ou de reliquats lors de la fermeture des fissures fœtales. Ils sont classés en trois grades :
les dermoïdes du limbe de grade I sont des lésions superficielles localisées au niveau du limbe et mesurant moins de 5 mm ;
les dermoïdes du limbe de grade II représentent des lésions plus larges recouvrant la plupart de la cornée et avec une extension profonde au niveau du stroma atteignant parfois jusqu’à la membrane de Descemet sans l’impliquer ;
le grade III est le plus rare avec des lésions très étendues de la cornée et pouvant atteindre jusqu’à l’épithélium pigmentaire de l’iris.
Les dermoïdes du limbe sont des lésions congénitales visibles dès la naissance. Selon son extension, la lésion peut conduire à une amblyopie ou un astigmatisme oblique aplatissant la cornée et revêtir un aspect inesthétique chez l’enfant. L’étude de Nevares rapporte que 76 % des dermoïdes du limbe sont situés en inférotemporal et 22 % en supérotemporal (fig. 11-42) [2]. L’étude histologique retrouve de 7 à 33 % de choristomes selon les études. Ils peuvent être associés à d’autres lésions oculaires telles que les staphylomes scléraux, les aniridies, les aphakies congénitales, les cataractes congénitales ou les microphtalmies.

Fig. 11-42 Dermoïde du limbe.
La transmission peut être génétique, soit : autosomique dominante, récessive, liée à l’X ou multifactorielle. Les dermoïdes du limbe peuvent être associés à divers syndromes et en particulier le syndrome de Goldenhar (fig. 11-43) et le syndrome de Franceschetti (ou syndrome de Treacher-Collins). Certains dermoïdes du limbe peuvent être associés à des nævi épidermiques qui doivent être surveillés. Les nævi sébacés doivent être identifiés, car ils peuvent donner lieu à des transformations malignes.

Fig. 11-43 Syndrome de Goldenhar.
La prise en charge est médico-chirurgicale.
La prise en charge médicale comporte la correction de l’astigmatisme induit et la rééducation de l’amblyopie.
Les études rapportent que 50 à 68 % des dermoïdes ne nécessitent qu’une exérèse chirurgicale simple [3]. L’indication est en général portée devant : la taille de la lésion ; l’astigmatisme induit avec éventuelle amblyopie difficile à rééduquer ; l’aspect esthétique. L’indication chirurgicale doit être discutée avec les parents en indiquant le risque potentiel de cicatrice. Un certain nombre de techniques chirurgicales ont été décrites dans la littérature depuis la simple exérèse jusqu’à la kératoplastie lamellaire ou transfixiante selon le grade de la lésion ou en faisant appel à une reconstruction par greffe de membrane amniotique en multicouche [4, 5]. Un bilan sera effectué sur la taille et la profondeur du dermoïde de façon à poser l’indication et la technique chirurgicale adaptée. Certains cas extrêmes de grade III, s’accompagnant de micro-ophtalmie, nécessitent une reconstruction du segment antérieur complexe ou parfois même une éviscération si le globe n’est pas fonctionnel. La prise en charge médico-chirurgicale vise à une récupération fonctionnelle réfractive et esthétique.
Ils sont habituellement localisés au niveau du canthus latéral et sont constitués de tissu adipeux. Classiquement, leur exérèse est pratiquée pour des raisons esthétiques. Il faut alors éviter les complications cicatricielles au niveau du muscle droit latéral et de la conjonctive.
Le syndrome auriculo-oculo-vertébral ou syndrome de Goldenhar est une anomalie de clivage au niveau du premier arc avec anomalie de migration des crêtes neurales. Il peut associer un dermoïde du limbe, un dermolipome, un colobome oculaire, des anomalies malformatives sévères du globe oculaire (microphtalmie, cryptophtalmie, etc.), des enchondromes prétragiens, des anomalies auriculaires et des anomalies vertébrales. Il peut aussi s’agir d’un syndrome de Goldenhar plus quand des anomalies cardiaques ou pulmonaires sont associées.
[1] Shields JA, Shields CL. Orbital cysts of childhood--classification, clinical features, and management. Surv Ophthalmol 2004 ; 49 : 281-99.
[2] Nevares RL, Mulliken JB, Robb RM. Ocular dermoids. Plast Reconstr Surg 1988 ; 82 : 959-64.
[3] Pirouzian A. Management of pediatric corneal limbal dermoids. Clin Ophthalmol 2013 ; 7 : 607-14.
[4] Burillon C, Duran L. Solid dermoids of the limbus and the cornea. Ophthalmologica 1997 ; 211 : 367-72.
[5] Mansour AM, Barber JC, Reinecke RD, Wang FM. Ocular choristomas. Surv Ophthalmol 1989 ; 33 : 339-58.