Pathologies immunes conjonctivosclérales et apparentées
J.-L. Metzinger, B. Wentworth, S.-D. Anesi, C.-S. Foster
La pemphigoïde oculaire cicatricielle est une maladie auto-immune chronique caractérisée par une inflammation progressive fibrosante de la conjonctive. Elle est souvent associée à des atteintes de la peau et d’autres muqueuses mettant en jeu parfois le pronostic vital. La maladie est spontanément potentiellement cécitante par ses lésions cornéennes. La mise en évidence par immunofluorescence directe ou technique immuno-enzymatique d’un dépôt immun linéaire sur la lame basale épithéliale signe le diagnostic de la maladie. Aucun traitement local n’est efficace. Les corticoïdes par voie systémique au long cours provoquent des effets secondaires inacceptables. Seuls les immunomodulateurs ont fait leurs preuves. Un diagnostic précoce reste cependant la meilleure garantie de succès thérapeutique.
La pemphigoïde oculaire cicatricielle (POC) est une maladie auto-immune chronique caractérisée par une inflammation progressive fibrosante de la conjonctive [1–3].
Elle appartient à un groupe hétérogène de maladies inflammatoires chroniques avec bulles sous-épithéliales appelé par les Anglo-Saxons mucous membrane pemphigoid (MMP), qui est souvent associé à une atteinte systémique et dont elle partage des caractéristiques physiopathologiques et cliniques. Le terme MMP est d’ailleurs souvent utilisé directement pour désigner non seulement la POC, mais aussi ses autres entités qui sont la pemphigoïde cicatricielle, la pemphigoïde buccale et la pemphigoïde bénigne des muqueuses (benign mucosal membrane pemphigoid) [1].
Les atteintes extra-oculaires les plus fréquentes de la MMP sont les muqueuses buccale, œsophagienne et laryngée, et la peau [3]. Les cicatrices fibreuses et rétractiles peuvent être source dans certaines localisations de complications graves voire mortelles [4]. En raison de son pronostic visuel spontané fréquemment sévère, notamment en cas d’atteinte bilatérale, la POC suscite un intérêt clinique certain non seulement auprès des ophtalmologistes, mais aussi d’autres spécialistes [2].
Jusqu’à présent aucun traitement local n’a fait la preuve de son efficacité pour stopper la progression de la fibrose conjonctivale de la POC ; les corticoïdes par voie systémique à hautes doses peuvent contrôler la maladie, mais au détriment d’un risque élevé de complications iatrogéniques à court et long terme [2]. Un traitement corticoïde en monothérapie au long cours de la POC est devenu inacceptable depuis que les immunomodulateurs par voie systémique ont prouvé leur efficacité [1, 2].
La gravité de la maladie et le manque de ressources thérapeutiques, une fois initié le processus de fibrose, justifient l’importance fondamentale d’un diagnostic précoce. Celui-ci est cependant rendu difficile à cause de la rareté relative de la maladie et l’absence de spécificité des symptômes au stade initial [5].
Le mot pemphigoïde tire ses racines du grec pemphix, qui signifie air, bouffée, gonflement ou vésicule. Selon Lever et Talbot, Hippocrate a été le premier à l’utiliser pour décrire certains états fébriles [2, 6]. C’est seulement au xviiie siècle que les médecins Koenig et Wichmann définissent les entités respectives de pemphigoïde avec atteinte systémique et de pemphigoïde strictement conjonctivale [2, 7, 8]. D’autres, dès 1768, assimilent la POC au pemphigus, et von Graefe est à l’origine de la première description clinique détaillée, à tous les stades de l’évolution [2, 9]. La distinction clinique et histopathologique entre pemphigoïde et pemphigus revient à un dermatologue, Lever, en 1953 [2, 10]. Malgré de nombreuses dénominations au fil du temps, la POC reste le terme le plus utilisé, même s’il prête à confusion puisque la majorité des patients ont aussi une atteinte systémique [1–3]. Les éléments clés du diagnostic de POC restent néanmoins constants : formation de bulles sous-épithéliales qui se rompent pour laisser des cicatrices cutanées et au niveau des muqueuses ; en immunopathologie, maladie auto-immune par hypersensibilité de type II selon la classification de Gell et Coombs avec dépôts linéaires d’immunoglobulines A (IgA), G (IgG) et/ou de complément 3 (C3) sur la membrane basale de peau et de la conjonctive [1]. La femme serait deux fois plus touchée que l’homme [1].
L’incidence de la POC est difficile à établir avec précision, mais elle est probablement sous-évaluée [2]. Dans la littérature ophtalmologique, elle se situe entre 1/20 000 et 1/46 000, avec des maximums de 1/12 000 et des minimums de 1/60 000 [1–3]. D’après les experts, les formes débutantes de la maladie échapperaient en partie aux statistiques [2]. La POC reste dans tous les cas une maladie rare. L’âge moyen auquel le diagnostic est fait est de 65 ans avec des extrêmes allant de 30 à 90 ans [1, 3]. Aucune prédilection raciale ou géographique n’a encore été rapportée [1, 5].
Bien que la pathogénie de la POC reste encore imparfaitement connue, il est clair que cette maladie résulte d’une dysrégulation immunitaire et répond à un mécanisme d’hypersensibilité de type II. Six auto-antigènes potentiels ont été identifiés au niveau de la membrane basale conjonctivale et de l’épiderme : les sous-unités α6/β4 de l’intégrine, la laminine 332, les antigènes BP180 et BP230 de la pemphigoïde bulleuse et le collagène de type VII. La diversité de ces cibles de la membrane basale laisse penser que la POC représente en fait un ensemble de mécanismes physiopathogéniques dont la résultante anatomo-pathologique serait identique [11]. Cependant l’auto-antigène de loin le plus important est la sous-unité β4 de l’intégrine α6β4 [12]. Plus précisément, c’est un domaine intracellulaire du peptide β4 situé au niveau des hémi-desmosomes des cellules basales épithéliales qui serait le site de liaison avec les auto-anticorps impliqués dans la maladie [12].
Le facteur déclenchant, chez un patient génétiquement prédisposé, peut être un médicament ou un microbe. Des clones de lymphocytes B fabriquent alors des auto-anticorps contre le peptide β4 auquel ils se lient pour induire ensuite l’activation du complément, le recrutement de cellules inflammatoires (lymphocytes, mastocytes et plasmocytes) et la libération de cytokines, avec pour résultante la formation de bulles sous-épithéliales [2]. Il s’ensuit alors une activation et une prolifération des fibroblastes qui produisent du néocollagène pourvoyeur de fibrose cicatricielle. Les conséquences en sont : une altération de l’épithélium cornéen ; des remaniements anatomiques de la conjonctive ; un dysfonctionnement meibomien ; un film lacrymal pathologique ; un xérosis et des lésions cornéennes par traumatisme mécanique par des cils ectopiques ; un trichiasis ou encore des marges palpébrales kératinisées.
Certains patients sont plus prédisposés que d’autres à développer une POC suivant l’hypothèse classique à « deux coups », selon laquelle le second coup est un facteur déclenchant environnemental (encadré 7-1).
• Étiologie auto-immune : pemphigoïde oculaire cicatricielle, syndrome de Stevens-Johnson, maladie du greffon contre l’hôte, sclérodermie, lupus érythémateux aigu disséminé, syndrome de Sjögren, maladie de Wegener, sarcoïdose.
• Étiologie conjonctivale : blépharoconjonctivite atopique, conjonctivite ligneuse, blépharoconjonctivite de la rosacée, les autres causes de conjonctivite infectieuse membraneuse.
• Infections :
■ bactéries : Corynebacterium diphtheriae, streptocoque β-hémolytique, Neisseria gonorrhea, Chlamydia trachomatis, Lymphogranuloma venerum ;
■ virales : adénovirus, conjonctivite à Herpes simplex.
• Iatrogénie médicamenteuse :
■ systémique : practolol, D-pénicillinamine ;
■ topique : épinéphrine, échothiophate iodide, pilocarpine, idoxuridine.
• Maladies systémiques bulleuses : syndrome de Lyell, épidermolyse bulleuse acquise, pemphigus vulgaris.
• Traumatismes : brûlures chimiques, brûlures thermiques, traumatismes mécaniques, radiations ionisantes, chirurgie conjonctivale, fistule carotido-caverneuse.
D’après Kirzhner M, Jakobiec FA. Ocular cicatricial pemphigoid : a review of clinical features, immunopathology, differential diagnosis, and current management. Seminars in Ophthalmology 2011 ; 26 : 270-7.
Les premiers haplotypes associés à la POC étaient les allèles HLA-DR4 et HLA-DQw3 [13]. Nos études de familles d’haplotypes DQ3 par analyse RFLP (restriction fragment length polymorphism, soit polymorphisme de longueur des fragments de restriction en français) ont démontré une association entre le gène HLA-DQb1*0301 et une plus grande susceptibilité à développer une POC ; cependant, d’autres études de séquençage génomique montrent que les séquences d’acides aminés des chaînes HLA-DQβ de patients atteints et de parents à eux sont identiques. Cela suppose que le véritable gène de susceptibilité à développer une POC n’est pas le HLA-DQb1*0301, mais plutôt un autre très proche de lui [14].
L’étude immuno-histopathologique des biopsies de conjonctive de POC met en évidence plusieurs éléments. Le dépôt immun linéaire en immunofluorescence ou immunoperoxidase d’IgG, C3 et parfois IgA et/ou IgM au niveau de la lamina lucida de la membrane basale est quasi pathognomonique de la POC et la différencie des autres pathologies pemphigoïdes [2, 3]. La présence simultanée d’IgG et IgA dans la membrane basale serait un élément associé à des formes plus sévères de la maladie [15]. Dans sa phase de début, on relève : plus de desmosomes et de matériel fibrillaire intra-épithélial ; moins d’hémidesmomes ; une métaplasie malpighienne de l’épithélium ; une diminution du nombre de cellules à mucus ; un infiltrat inflammatoire cellulaire lymphoplasmocytaire ; une fibrose de la substantia propria.
La manifestation oculaire initiale de la POC est une conjonctivite unilatérale d’évolution chronique [2]. Les symptômes consistent principalement en une irritation oculaire, une sensation de brûlure, un larmoiement, une sécrétion filamenteuse de mucus dans les culs-de-sac conjonctivaux, une rougeur conjonctivale et une kératopathie ponctuée superficielle irrégulière, mais très rarement des vésicules (fig. 7-1) [1, 2]. Certains patients peuvent rester asymptomatiques, même aux stades avancés de la maladie [1]. Après des épisodes récurrents d’exacerbations inflammatoires se produit parfois alors une bilatéralisation du processus avec progression à différents stades et degrés de sévérité de la maladie [1]. On peut diagnostiquer alors une fibrose sous-épithéliale sous forme de fines stries blanches au niveau des conjonctives tarsales inférieures et/ou supérieures [2].
Pour suivre la progression de la maladie, instaurer son traitement et en évaluer l’efficacité, et établir son pronostic, il est nécessaire d’en connaître la classification des stades évolutifs [3] :
le stade I de la POC est caractérisé par la conjonctivite chronique avec fibrose sous-épithéliale visible sous la forme de stries blanches au niveau des conjonctives tarsales (fig. 7-2) [2] ;
le stade II correspond au comblement du fornix inférieur (fig. 7-3) [2] ;
le stade III se distingue par la formation de symblépharons (fig. 7-4) qui sont associés à divers degrés de kératopathie, néovascularisation cornéenne, trichiasis, dystichiasis, kératinisation des conjonctives palpébrales (fig. 7-5) et déficience lacrymale [2] ;
le stade IV, terminal, est marqué par l’ankyloblépharon, un syndrome de sécheresse oculaire sévère et un xérosis (fig. 7-6) [2].
Pour apprécier au mieux la progression souvent irrégulière de la maladie, une iconographie est recommandée [3].
Environ la moitié des patients souffrant de POC ont ou auront une atteinte extra-oculaire [1]. À l’interrogatoire, on peut ainsi retrouver de manière diversement associée une atteinte de la peau, du cuir chevelu, des muqueuses buccales et nasales, du pharynx, du larynx, de l’œsophage, du vagin, de l’urètre, et/ou de l’anus [2]. La prise en charge de la maladie doit être alors multidisciplinaire. Si la fibrose dans certaines localisations peut ne pas évoluer, certaines lésions comme les ulcérations des muqueuses n’en sont pas moins très invalidantes [2]. Les extractions dentaires extensives sont de règle en cas de gingivite desquamative et des pertes osseuses sont des séquelles possibles [2]. Les lésions cutanées sont des vésicules ou bulles récidivantes guérissant sans séquelles, ou des plaques érythémateuses prurigineuses laissant des cicatrices [2]. Une rétraction progressive des cicatrices est visible au niveau des muqueuses vaginale, urétrale, et anale ; elle peut mettre en jeu le pronostic vital lorsqu’elle touche l’œsophage ou la trachée [2]. Une asphyxie par obstruction, par bol alimentaire ou accumulation de mucus, et entraînant la mort a aussi été rapportée chez des patients avec POC [2].

Fig. 7-1 Conjonctive inflammatoire avec vésicules.
La suspicion clinique de POC doit rester forte en raison de la progression rapide de la maladie et de son pronostic d’autant plus sévère que le diagnostic est retardé.

Fig. 7-2 POC stade I.
Noter les stries fibreuses sous-épithéliales conjonctivales.
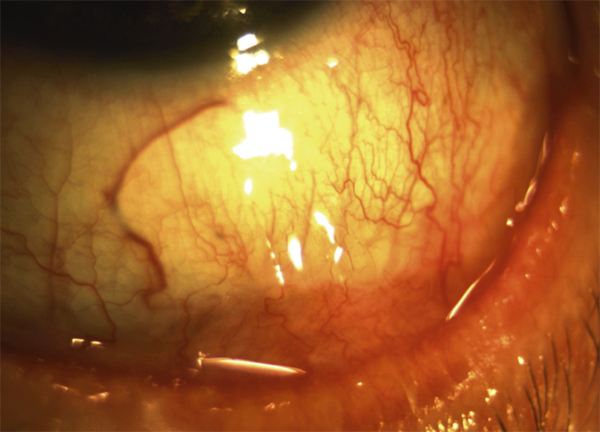
Fig. 7-3 POC stade II : comblement du fornix inférieur.
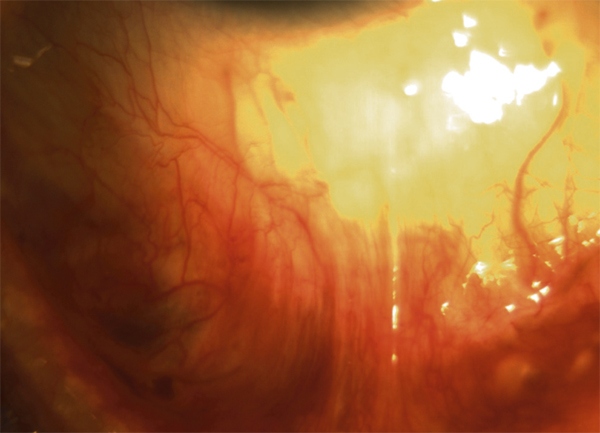
Fig. 7-4 POC stade III : symblépharons.

Fig. 7-5 POC stade III : plaque de kératinisation sur la conjonctive palpébrale inférieure.
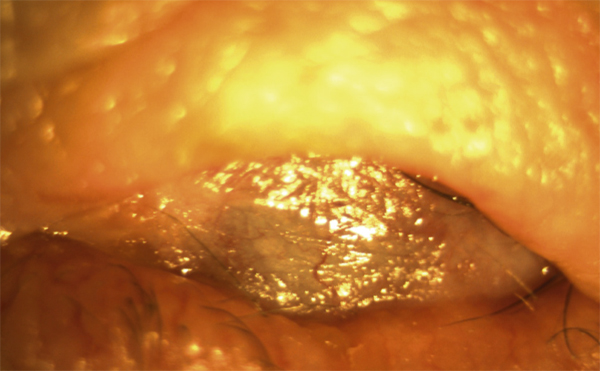
Fig. 7-6 POC stade IV ou terminal.
Le fornix inférieur a disparu pour être remplacé par un ankyloblépharon, et la surface oculaire est entièrement kératinisée (xérosis).
Il est important de différencier la POC des autres causes de conjonctivites fibrosantes (voir encadré 7-1) qui sont d’autant plus nombreuses que cette maladie a des présentations diverses et variées. Au stade de début, la POC peut passer pour un simple syndrome de sécheresse oculaire car les signes de fibrose et le caractère évolutif de la maladie sont absents. L’existence d’une conjonctivite folliculaire aux stades précoces pourrait aussi faire penser à une multitude d’étiologies telles qu’une conjonctivite infectieuse, allergique ou iatrogénique provoquée par des médicaments, ou d’autres processus inflammatoires systémiques comme le syndrome de Gougerot-Sjögren, une sarcoïdose ou un lupus érythémateux disséminé. Cela explique les retards et erreurs diagnostiques habituels aux premiers stades de la maladie. La rosacée oculaire et les formes avancées de blépharo-kératoconjonctivites atopiques peuvent simuler une POC inflammatoire à la fois à son début et dans ses formes avancées avec fibrose. Une association de ces maladies à la POC n’est pas exceptionnelle non plus.
En cas de fibrose chronique, le diagnostic différentiel peut se faire avec quelques maladies bulleuses rares apparentées à la POC, comme la maladie linéaire à IgA, l’épidermolyse bulleuse acquise et la pemphigoïde bulleuse ; elles ont toutes un dépôt linéaire immun au niveau de la membrane basale épithéliale visible en immunofluorescence directe [16]. Cependant la maladie linéaire à IgA ne progresse habituellement pas, et l’épidermolyse bulleuse acquise touche rarement les yeux [3]. Le lichen plan pemphigoïde peut entraîner une fibrose aussi importante que la POC, mais l’aspect chevelu des dépôts immuns de fibrinogène en immunofluorescence directe ainsi que les images de duplication en microscopie électronique l’en distinguent ; il faut rappeler que la présence de fibrinogène est normale sur la membrane basale de la conjonctive contrairement à la muqueuse buccale [16].
Les causes infectieuses de conjonctivite cicatricielle sont parfois diagnostiquées grâce aux techniques de culture [3]. Une conjonctivite membraneuse peut se voir avec l’adénovirus et le virus herpès, et des bactéries comme le streptocoque β-hémolytique, le gonocoque et le bacille de la diphtérie [3]. Les chlamydioses dont fait partie le trachome sont aussi responsables de conjonctivite cicatricielle, mais l’inflammation débute habituellement sur la conjonctive palpébrale supérieure [3].
La conjonctivite fibrosante iatrogénique peut aussi se présenter comme la POC, mais sa topographie prédomine habituellement dans le fornix inférieur et elle est associée à un antécédent de prise prolongée de collyres ; la conjonctivite de la POC à son début est plus marquée au canthus et sur la conjonctive tarsale supérieure [17, 18]. Les fibroses conjonctivales chroniques induites par des médicaments à usage systémique sont beaucoup plus rares depuis que le médicament le plus incriminé (practolol) a été retiré du marché [3]. Une bonne enquête pharmacologique doit aider à éliminer ces causes iatrogéniques [3]. Un interrogatoire et un bilan systémique précis élimineront facilement d’autres causes de fibrose conjonctivale telles que la maladie du greffon contre hôte, les syndromes de Lyell et Stevens-Johnson, les radiations ionisantes et les traumatismes chirurgicaux et non chirurgicaux.
Il est crucial d’établir avec certitude le diagnostic de POC afin de mettre en œuvre le traitement pour préserver la vision. Lors de la première conférence internationale de consensus sur les MMP, des critères diagnostiques à la fois cliniques et histopathologiques ont été définis [19]. Un patient est considéré comme atteint de MMP s’il est porteur d’une maladie bulleuse inflammatoire chronique intéressant une ou plusieurs muqueuses avec ou sans atteinte cutanée ou cicatrice [20]. En immunofluorescence directe, la biopsie de la conjonctive atteinte doit mettre en évidence un élément pathognomonique pour le diagnostic de POC : le dépôt linéaire immun d’IgG, C3 et/ou IgA sur la membrane basale épithéliale (fig. 7-7) [19]. Il est de plus possible de détecter dans le sérum des anticorps circulants dirigés contre la membrane basale épithéliale de la conjonctive grâce à des tests radio-immunologiques très sensibles [11]. Des anticorps circulants contre l’épithélium conjonctival ainsi que des anticorps antinucléaires sont aussi parfois retrouvés [21, 22].
Par contre, une biopsie négative n’élimine pas le diagnostic de POC ; en cas de forte présomption basée sur la clinique, les pièces de biopsie éventuellement répétée peuvent être traitées avec des techniques de marquage plus sensibles telles l’immunoperoxidase [3]. Celle-ci utilise le complexe avidine–biotine et augmente le taux de positivité des biopsies conjonctivales de 52 à 83 % par rapport à l’immunofluorescence [3, 23]1. Le prélèvement doit être idéalement réalisé à la limite de la conjonctive enflammée (avec ou sans fibrose) [3]. Il est aussi impératif de porter immédiatement les tissus frais pour analyse ; dans le cas contraire, les tissus peuvent être conservés jusqu’à 15 jours dans un fixateur Zeus ou Michel [2, 3].
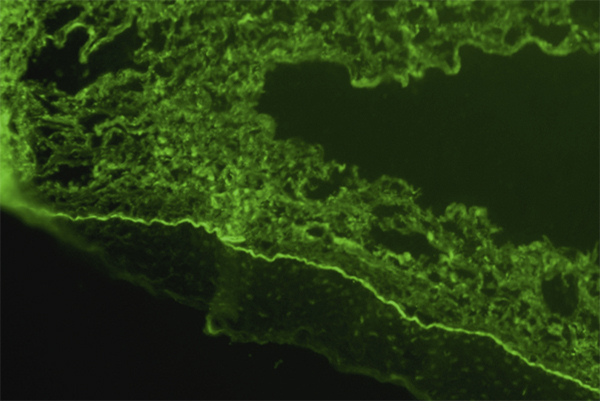
Fig. 7-7 Immunofluorescence sur une biopsie conjonctivale d’un patient suspect d’avoir une POC.
La bande verte fluorescente en bas représente un dépôt d’IgG humaines sur la membrane basale épithéliale conjonctivale qui confirme le diagnostic de POC.
Le but final du traitement est de supprimer l’inflammation et de guérir le patient de sa pathologie en prévenant au mieux les séquelles cicatricielles [1, 21]. Le choix du traitement doit être adapté à la gravité et/ou au stade évolutif de la maladie et en fonction du nombre et du type de sites touchés ; cependant les patients avec atteinte oculaire sont systématiquement considérés comme à « haut risque » et requièrent un traitement par voie systémique [5, 19, 23]. L’ophtalmologiste doit prendre en compte les atteintes extra-oculaires éventuelles lorsqu’il pose ses indications thérapeutiques et prescrit son traitement [3]. Le bilan médical initial et au cours du suivi est d’une haute importance [3].
Il n’est jamais superflu de répéter que les traitements locaux à base de corticoïdes et de substituts des larmes sont totalement inefficaces pour ralentir la progression de la POC [2]. Les collyres lubrifiants sans conservateurs, le sérum autologue et les bouchons lacrymaux ne sont pas pour autant inutiles car ils contribuent à soulager le syndrome de sécheresse oculaire dont se plaignent très souvent les patients [1]. Les injections sous-conjonctivales de corticoïdes peuvent être bénéfiques temporairement mais pas à long terme et favorisent de surcroît la cataracte [2]. Une hygiène des paupières méticuleuse, des compresses chaudes et la tétracycline orale ou ses dérivés pour traiter une blépharite peuvent constituer des adjuvants thérapeutiques annexes à visée symptomatique [1].
Les corticoïdes par voie systémique en monothérapie ne sont pas non plus indiqués. Bien au contraire, ils garantissent presque inéluctablement des effets secondaires s’ils sont prescrits au long cours alors qu’ils n’assurent pas un contrôle stable de la maladie ; tout juste sont-ils capables de maîtriser l’inflammation conjonctivale mais pas la progression de la fibrose [2–4]. Ces complications iatrogènes sont l’hyperglycémie, l’hypertension artérielle, l’hyperlipidémie, l’ostéoporose, les ulcères gastriques, des infections sévères et des troubles psychologiques pouvant aller jusqu’à une psychose [5]. Dans une étude sur 81 patients diagnostiqués POC, il a été démontré qu’une posologie quotidienne de 40 mg de prednisone était nécessaire pour stopper la progression de la maladie ; malgré ces fortes doses d’entretien, 17 patients sur 62 qui avaient une atteinte oculaire inflammatoire active sont devenus aveugles pendant la période de traitement [2, 25]. Des complications iatrogènes graves en rapport avec les corticoïdes étaient aussi rapportées chez trois patients : un mort par pneumonie, un cas d’hémorragie cérébrale et un cas d’hémorragie digestive, tous deux à cause d’une hypertension artérielle due au médicament [25].
Les corticoïdes par voie systémique peuvent néanmoins être utilisés comme adjuvants thérapeutiques afin de juguler provisoirement l’inflammation, le temps que le traitement immunomodulateur systémique fasse son effet [3]. Une dose de 1 à 1,5 mg/kg/jour représente la posologie initiale habituelle, associée à une surveillance régulière des effets secondaires et une supplémentation recommandée en calcium et vitamine D [26]. Les modalités de sortie du traitement dépendent de la réponse clinique au traitement initié [26]. La dapsone par voie systémique constitue aussi un complément utile au début dans les formes graves, à la dose initiale de 25–50 mg/jour jusqu’à la rémission clinique [2, 4, 26]. Elle requiert une surveillance de l’hémoglobine à la recherche d’une anémie 1 semaine après le début du traitement et 1 semaine après toute augmentation de la posologie, car les effets secondaires peuvent être sérieux, incluant surtout l’anémie hémolytique, l’agranulocytose et une anémie par aplasie médullaire [3, 5]. Les doses efficaces quotidiennes habituelles sont de 100 à 200 mg ; l’augmentation de la posologie se fait par paliers de 25 mg tous les 7 jours [5].
Les traitements par immunosuppresseurs ne sont classiquement préconisés qu’à partir du stade 2 d’une maladie évolutive [19]. Pour un résultat optimal, la prise en charge du patient doit être si possible multidisciplinaire, faisant appel à un oncologue ou un rhumatologue [3]. Avant la mise en route du traitement, des tests sanguins sont nécessaires pour éliminer une contre-indication et servir de référence pour la surveillance ultérieure. Ils comportent les tests de fonction hépatique et rénale, une numération formule sanguine complète et la recherche d’un déficit en glucose-6-phosphate déshydrogénase [3]. Le patient doit recevoir une information complète relative à toutes les options de traitements immunosuppresseurs. Il faudrait aussi que les soignants expliquent que l’immunomodulation est impérative pour empêcher l’inflammation de progresser, mais ne peut faire disparaître les lésions déjà constituées [3].
Comme pour toute autre maladie inflammatoire oculaire, le traitement de la POC est délivré de manière graduelle, avec des options thérapeutiques immunosuppressives de plus en plus agressives en fonction des résistances ou échecs précédents. En première ligne, les traitements les plus utilisés sont le mycophénolate mofétil, le méthotrexate, l’azathioprine et la ciclosporine, isolés ou diversement associés entre eux. Les produits issus de la biotechnologie recombinante de l’ADN, les agents alkylants et les autres thérapeutiques comme les immunoglobulines par voie intraveineuse représentent des options plus agressives pour les patients atteints de POC. Ces traitements posent des problèmes d’administration et de compliance lorsque les patients souvent âgés sont adressés tardivement et vivent seuls et/ou loin ; il n’est pas rare que la maladie soit découverte à un stade avancé et requière un traitement agressif et permanent [1]. L’éducation du patient est en cela fondamentale pour la compliance médicale.
Au total, très peu d’études prospectives ont été conduites à bien pour évaluer tous ces types de traitements de la POC ; les cliniciens doivent se reposer sur les données de petites séries rétrospectives. Le mycophénolate mofétil s’est révélé très efficace en association avec les corticoïdes par voie générale dans quatre publications de séries de cas [27–30]. Les effets secondaires étaient de surcroît peu importants [27]. L’étude SITE (systemic immunosuppressive therapy for eye diseases) a mis en évidence l’efficacité du mycophénolate mofétil dans une cohorte de 18 patients avec POC ; l’inflammation était contrôlée avec un recul de 12 mois chez 71 % des patients [31]. Comme elle, d’autres études ont montré que cette molécule était pourvoyeuse de taux d’effets secondaires moindres par rapport aux autres immunomodulateurs classiques [31–32].
L’efficacité du méthotrexate dans la POC a été évaluée par McCluskey et al. ; 10 sur 12 patients ont répondu remarquablement bien à un traitement de 15 mois avec une disparition de la conjonctivite et/ou un contrôle total de la maladie avec un recul moyen supérieur à 30 mois [33]. Vingt-quatre pour cent des patients dans cette série ont eu des effets secondaires imposant la suspension seulement temporaire du traitement (troubles digestifs, fatigue, alopécie) [32]. Dans l’étude SITE incluant 109 yeux de 58 patients traités par méthotrexate, 87 % des patients n’avaient plus d’inflammation avec un recul de 12 mois [34]. L’azathioprine dans la POC a fait l’objet d’une évaluation par Saw et al. et le groupe d’étude de cohortes SITE ; tous ont conclu que l’azathioprine permettait de contrôler la progression de la POC [31, 34]. Mais le taux de succès était inférieur à celui du mycophénolate mofétil et du méthotrexate, et les effets secondaires étaient plus fréquents, à type de troubles digestifs, céphalées, sensation de malaise, vertiges, élévation des transaminases hépatiques et aplasie médullaire [23, 32, 35]. Quant à la ciclosporine, elle ne semble pas aussi efficace dans le traitement au long cours de la POC que dans celui d’autres maladies inflammatoires oculaires ; pour l’heure et au regard du trop peu d’études sur le sujet, elle ne fait pas partie des options thérapeutiques de la POC [36].
Le cyclophosphamide, un agent alkylant, est utilisé avec succès depuis quelques décennies dans le traitement des POC modérées à sévères [5]. Seul ou associé aux corticoïdes par voie générale, il représente une option thérapeutique agressive qui permet de contrôler rapidement la progression de la maladie chez les patients réfractaires [5]. Comparé à la dapsone, la sulfapyridine, l’azathioprine et le mycophénolate mofétil, le cyclophosphamide s’avère le plus efficace (environ 91 % de taux de succès) [32]. L’étude de Thorne et al. a révélé que le cyclophosphamide associé aux corticoïdes par voie orale augmentait les chances de rémission oculaire dans la POC par un facteur de 8,5 par rapport à la dapsone, les corticoïdes en monothérapie et les immunomodulateurs classiques [37]. Ces bons chiffres ne justifient pas pour autant que tout patient reçoive du cyclophosphamide ; en effet, ce médicament n’est pas anodin et doit faire l’objet d’une surveillance toute particulière si on ne veut pas qu’il provoque plus de mal que de bien [2, 38, 39]. Dans les deux études précédentes, une proportion significative de patients était obligée d’arrêter le traitement à cause d’effets secondaires intolérables [32, 37]. Ceux-ci incluent un risque accru de développement de cancers, une hépatotoxicité, une leucopénie, une anémie, une alopécie, une anovulation, une azoospermie, une cystite hémorragique et une aplasie myéloïde [3, 32]. Le cyclophosphamide est prescrit pour une période d’un an par voie orale à la dose de 50 à 200 mg/jour, ou par voie intraveineuse à la dose de 1 à 2,5 mg/kg/jour, ou par bolus intraveineux à la dose mensuelle de 0,5 à 1 g/m2 [26]. Des essais contrôlés randomisés sur de larges populations comparant le cyclophosphamide avec les autres alternatives détaillées plus haut sont nécessaires pour appuyer ces recommandations.
Des études plus récentes semblent indiquer que le rituximab, seul ou associé à d’autres médicaments (dapsone, immunoglobulines par voie intraveineuse), est une alternative thérapeutique valable pour les patients avec POC modérée à sévère et réfractaires aux autres traitements [23]. Le rituximab est un anticorps monoclonal chimérique dirigé contre le CD20 ; il est administré à la dose initiale hebdomadaire de 375 mg/m2 pendant 8 semaines ; puis le traitement est reconduit à la même posologie pendant les 4 mois suivants [23]. Une étude rapporte que six patients ne répondant pas favorablement aux immunoglobulines intraveineuses ont eu une stabilisation de leur maladie grâce au rituximab avec un suivi de 11 mois après arrêt du traitement [40]. Une autre publication sur 25 patients révèle que dix patients sur dix avec atteinte oculaire et traités avec une association rituximab–dapsone ont totalement guéri de leurs yeux [41]. Les risques associés au rituximab sont les infections sévères et justifient en cela d’une surveillance étroite par l’équipe médicale [23]. Le coût du médicament et les contraintes liées à son administration, qui plus est en association avec les immunoglobulines par voie intraveineuse, représentent les principaux freins à sa prescription, malgré sa remarquable efficacité et sa faible toxicité. Davantage de recherches sont encore nécessaires pour préciser la place du rituximab dans le traitement de la POC.
Les immunoglobulines par voie intraveineuse sont une autre option thérapeutique pour les patients répondant mal aux traitements conventionnels. Des petites séries de cas et des études rétrospectives indiquent que des travaux complémentaires sont requis afin d’améliorer les connaissances quant à l’utilisation de ce traitement [26]. Les doses recommandées sont de 2 à 3 g/kg, typiquement administrées sur une période de 3 à 5 jours toutes les 4 semaines [26]. Des manifestations d’intolérance lors de l’administration du produit sont possibles, sous la forme de maux de tête, rougeur subite, nausées, frissons, tachycardie, hypotension artérielle et sifflement respiratoire. C’est pourquoi une prémédication et un monitoring sont nécessaires [26]. En plus du bilan de laboratoire standard préalable à la délivrance du médicament, il faut aussi éliminer un syndrome d’immunodéficience acquise (sida) et une hépatite [26].
La chirurgie intervient pour traiter un trichiasis, un dystichiasis, un entropion ou une lagophtalmie, mais seulement après contrôle de l’inflammation depuis au moins 3 mois [3]. Cette règle est fondamentale au risque de voir la chirurgie aggraver la POC [3].
Malheureusement, le pronostic est très imprévisible à cause de la variabilité des réponses des patients aux différents traitements [5]. Il y a une corrélation entre le degré de morbidité de la maladie et la rapidité avec laquelle elle a été reconnue et le traitement a été instauré ; une étude a montré que le diagnostic de POC était fait à un stade avancé (stade III ou plus) jusque dans 60 % des cas [1]. L’inflammation oculaire peut être jugulée dans la majorité des cas grâce un traitement approprié, précoce et agressif [2].
Les rechutes et phases de progression sont communes, en particulier chez les patients qui ont des atteintes multiloculaires. Une exacerbation des signes subjectifs et objectifs demande à ce que le traitement soit réadapté si nécessaire. La fibrose cicatricielle peut constituer une complication significative quand elle entraîne des rétractions tissulaires [5].
La POC est une maladie chronique délétère qui peut compromettre gravement la vision si elle n’est pas traitée. Elle est insidieuse et malgré sa dénomination, elle a des implications systémiques. Les progrès récents dans les techniques de diagnostic et les traitements ont aidé à une meilleure connaissance et prise en charge de la maladie. Au fil des progrès de la recherche dans les maladies inflammatoires oculaires, le nombre des options thérapeutiques augmente et cela contribue à un meilleur pronostic à court et long terme. Les innovations à venir vont renforcer les chances de préserver la vue alors qu’auparavant la cécité était inévitable.
[1] Al-Ghoul A, Kim G, Dhaliwal DK. Ocular cicatricial pemphigoid/Mucous membrane pemphigoid. In Yanoff M, Duker JS. Ed. Ophthalmology. Missouri : Elsevier ; 2008, p. 410.
[2] Foster CS. Cicatricial pemphigoid. Trans Am Ophthalmol Soc 1986 ; 84 : 527-663.
[3] Kirzhner M, Jakobiec FA. Ocular cicatricial pemphigoid : a review of clinical features, immunopathology, differential diagnosis, and current management. Semin Ophthalmol 2011 ; 26 : 270-7.
[4] Miserocchi E, Baltatzis S, Roque MR, et al. The effect of treatment and its related side effects in patients with severe ocular cicatricial pemphigoid. Ophthalmology 2002 ; 109 : 111-8.
[5] Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am 2013 ; 57 : 611-30.
[6] Lever WF, Talbott JH. Pemphigus : a historical study. Arch Dermatol Syph 1942 ; 46 : 800-23.
[7] Koenig S. Lettre contenant plusieurs symptômes étrangers et surprenants. Par F. Planque. Bibliothèque Choisie de Médecine 1748-1770 ; 7 : 577.
[8] Wichmann JE. Ideen zur Diagnostik. Helwig Hannover 1793 ; 1 : 89.
[9] von Graefe A. Bericht Deutsche Ophthalmol Gesell 1879 ; 12 : 234.
[10] Lever WF. Pemphigus. Medicine 1953 ; 32 : 1-123.
[11] Foster CS. Cicatricial Pemphigoid. Trans Am Ophthalmol Soc 1986 ; 84 : 527-663.
[12] Tyagi S, Bhol K, Natarajan K, et al. Ocular cicatricial pemphigoid antigen : partial sequence and biochemical characterization. Proc Natl Acad Sc USA 1996 ; 93 : 14714-9.
[13] Ahmed AR, Foster CS, Zaltas M, et al. Association of DQW7 (DQB1*0301) with ocular cicatricial pemphigoid. Proc Natl Acad Sc USA 1991 ; 88 : 11579-82.
[14] Haider N, Neuman R, Foster CS, et al. Report on the sequence of DQB1*0301 gene in ocular cicatricial pemphigoid patients. Curr Eye Res 1992 ; 11 : 1233-8.
[15] Setterfield J, Shirlaw PJ, Kerr-Muir M, et al. Mucous membrane pemphigoid : a dual circulating antibody response with IgG and IgA signifies a more severe and persistent disease. Br J Dermatol 1998 ; 138 : 602-10.
[16] Akpek EK, Ilhan-Sarac O. Cicatrizing conjunctivitis. The cornea : scientific foundations and clinical practice. 4th ed. Philadelphia : Smolin and Thoft ; 2005, p. 477-502.
[17] Schwab IR, Linberg JV, Gioia VM, et al. Foreshortening of the inferior conjunctival fornix associated with chronic glaucoma medications. Ophthalmology 1992 ; 99 : 197-202.
[18] Wright P. Enigma of ocular cicatricial pemphigoid : a comparative study of clinical and immunological findings. Trans Ophthalmol Soc UK 1979 ; 99 : 141-5.
[19] Chan LS, Ahmed AR, Anhalt HI, et al. The first international consensus on mucous membrane pemphigoid : definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002 ; 138 : 370-9.
[20] Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013 ; 381 : 320-32.
[21] Waltman SR, Yarian D. Circulating antibodies in ocular pemphigoid. Am J Ophthalmol 1974 ; 77 : 891-4.
[22] Mondino BJ, Brown SI, Rabin BS. Autoimmune phenomena of the external eye. Ophthalmology 1978 ; 85 : 801-17.
[23] Sobolewska B, Deuter C, Zierhut M. Current medical treatment of ocular mucous membrane pemphigoid. Ocul Surf 2013 ; 11 : 259-66.
[24] Robin H, Hoang-Xuan T, Prisant O, et al. Immunoelectron microscopic study of the conjunctiva in cicatricial pemphigoid. Graefes Arch Clin Exp Ophthalmol 1999 ; 237 : 39-44.
[25] Hardy KM, Perry HO, Pingree GC, et al. Benign mucous membrane pemphigoid. Arch Dermatol 1971 ; 104 : 467-75.
[26] Neff AG, Turner M, Mutasim DF. Treatment strategies in mucous membrane pemphigoid. Ther Clin Risk Manag 2008 ; 4 : 617-26.
[27] Staines K, Hampton PJ. Treatment of mucous membrane pemphigoid with the combination of mycophenolate mofetil, dapsone, and prednisolone : a case series. Oral Surg Oral Med Oral Pathol Oral Radiol 2012 ; 114 : e49-56.
[28] Doycheva D, Deuter C, Blumenstock G, et al. Long-term results of therapy with mycophenolate mofetil in ocular mucous membrane pemphigoid. Oc Immunol Inflamm 2011 ; 19 : 431-8.
[29] Rallis E, Anyfantakis V. Coexistent psoriasis and bullous pemphigoid responding to mycophenolate mofetil monotherapy. Skinmed 2008 ; 7 : 101-2.
[30] Nottage JM, Hammersmith KM, Murchison AP, et al. Treatment of mucous membrane pemphigoid with mycophenolate mofetil. Cornea 2013 ; 32 : 810-5.
[31] Daniel E, Thorne JE, Newcomb CW, et al. Mycophenolate mofetil for ocular inflammation. Am J Ophthalmol 2010 ; 149 : 423-32.
[32] Saw VP, Dart JK, Rauz S, et al. Immunosuppressive therapy for ocular mucous membrane pemphigoid. Strategies and outcomes. Ophthalmology 2008 ; 115 : 253-61.
[33] McCluskey P, Chang JH, Singh R, et al. Methotrexate therapy for ocular cicatricial pemphigoid. Ophthalmology 2004 ; 111 : 796-801.
[34] Gangaputra S, Newcomb CW, Liesegang TL, et al. Methotrexate for ocular inflammatory diseases. Ophthalmology 2009 ; 116 : 2188-98.
[35] Pasadhika S, Kempen JH, Newcomb CW, et al. Azathioprine for ocular inflammatory diseases. Am J Ophthalmol 2009 ; 148 : 500-9.
[36] Foster CS, Neumann R, Tauber J. Long-term results of systemic chemotherapy for ocular cicatricial pemphigoid. Doc Ophthalmol 1992 ; 82 : 223-9.
[37] Thorne JE, Woreta FA, Jabs DA, et al. Treatment of ocular mucous membrane pemphigoid with immunosuppressive drug therapy. Ophthalmology 2008 ; 115 : 2146-52.
[38] Elder MJ, Lightman S, Dart JK. Role of cyclophosphamide and high dose steroid in ocular cicatricial pemphigoid. Br J Ophthalmol 1995 ; 79 : 264-6.
[39] Tauber J, Sainz de la Maza M, Foster CS. Systemic chemotherapy for ocular cicatricial pemphigoid. Cornea 1991 ; 10 : 185-95.
[40] Foster CS, Chang PY, Ahmed AR, et al. Combination of rituximab and intravenous immunoglobulin for recalcitrant ocular cicatricial pemphigoid. Ophthlamology 2010 ; 117 : 861-9.
[41] Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol 2011 ; 147 : 843-9.
T. Hoang-Xuan
Les syndromes de Stevens-Johnson et de Lyell peuvent être réunis sous une même entité nosologique clinique et immunopathogénique. Ils désignent une éruption bulleuse mucocutanée aiguë de gravité très variable affectant l’adulte jeune, pouvant intéresser toutes les muqueuses dont la conjonctive. Il existerait une certaine prédisposition génétique. Les facteurs déclenchants habituels sont des agents infectieux – le Mycoplasma pneumoniae et le virus Herpes simplex hominis – et des médicaments, selon un mécanisme probablement immuno-allergique. Dans les formes dites majeures, le pronostic vital est mis en jeu. Les complications oculaires chroniques faisant suite à la conjonctivite aiguë du début sont fréquentes et parfois très sévères. Elles sont en relation avec la fibrose cicatricielle conjonctivale qui est directement ou indirectement responsable de symblépharons, ankyloblépharon, entropion–trichiasis, sécheresse oculaire, destruction des cellules souches limbiques, ulcères cornéens chroniques, abcès de la cornée et xérosis. Le traitement général des formes aiguës relève d’une prise en charge en unité de soins intensifs. Les médicaments soupçonnés doivent être retirés. Le traitement oculaire lors de la phase aiguë repose essentiellement sur la lubrification des yeux et une corticothérapie locale. En cas de persistance d’une inflammation sévère ou d’une récidive inflammatoire résistant aux anti-inflammatoires locaux, une immunosuppression systémique similaire à celle des maladies bulleuses auto-immunes est indiquée. Le traitement chirurgical vise à : aider à la cicatrisation avec les greffes de membrane amniotique, de cellules souches limbiques ou de muqueuse buccale transdifférenciée in vitro ; reconstruire l’architecture conjonctivopalpébrale ; restaurer la transparence avec les greffes cornéennes lamellaire ou transfixiante. La kératoprothèse ou l’ostéo-odontoprothèse sont seulement réservées aux formes très sévères de xérosis.
Les syndromes de Stevens-Johnson et de Lyell étaient encore décrits une trentaine d’années auparavant comme deux entités nosologiques cliniques et immunopathogéniques différentes, mais il existe de très nombreuses formes frontières rendant impossible cette distinction.
Les syndromes de Stevens-Johnson et Lyell désignent une éruption bulleuse mucocutanée aiguë intéressant très souvent peu ou prou la conjonctive. On en retrouve une première description en 1866 par Hebra qui donne le nom d’erythema exudativum multiforme à une entité associant conjonctivite purulente, stomatite et érythème polymorphe [1]. En 1922, Stevens et Johnson rapportent un syndrome d’ectodermose érosive pluri-orificielle qui comportait un érythème polymorphe et des lésions muqueuses sévères [2]. Enfin, Lyell introduit en 1956 le terme de toxic epidermal necrolysis pour décrire quatre cas de patients atteints de larges pertes cutanées associées à une atteinte des muqueuses [3]. En 1993, une conférence de consensus anglo-saxonne distingue trois entités, l’erythema multiforme minor, l’erythema multiforme major et la toxic epidermal necrolysis [4], qui correspondent respectivement aux expressions françaises d’érythème polymorphe, de syndrome de Stevens-Johnson et de syndrome de Lyell ou nécrose épidermique toxique. Mais l’érythème polymorphe, caractérisé par une atteinte cutanée isolée ou associée à au plus une lésion muqueuse qui est très rarement la conjonctive, est maintenant intégré au syndrome de Stevens-Johnson qui possède ainsi plusieurs degrés de sévérité. Certaines lésions cutanées seraient aussi plus caractéristiques du syndrome de Stevens-Johnson, et les muqueuses, dont surtout la conjonctive, peuvent être le siège d’une inflammation persistante ou récidivante [5]. Dans sa forme grave, comme le syndrome de Lyell, il touche la peau et plusieurs muqueuses dont la conjonctive qui se fibrose. Longtemps l’expression de syndrome de Lyell a été réservée aux formes les plus sévères d’emblée où le décollement bulleux cutané est très étendu, comparable à celui des grands brûlés, et dans lesquelles, une fois les cicatrices muqueuses constituées, il n’est plus constaté d’évolution inflammatoire à leur niveau. En fait, la distinction entre syndrome de Stevens-Johnson et syndrome de Lyell est très artificielle, souvent difficile et parfois impossible. C’est la raison pour laquelle il serait plus judicieux d’assimiler ces mucodermatoses bulleuses aiguës sous la même appellation « syndrome de Stevens-Johnson/Lyell ».
Le syndrome de Stevens-Johnson/Lyell est rare. Aux États-Unis, l’incidence cumulée de l’érythème polymorphe, du syndrome de Stevens-Johnson et du syndrome de Lyell est de 4,2 par million de personnes et par an. L’incidence du seul syndrome de Lyell est de 0,5 par million de personnes et par an [6]. En France, elle est de 1,2 à 1,3 par million de personnes et par an [7]. Ces dermatoses bulleuses auto-immunes surviennent plus souvent chez les patients immunodéprimés, particulièrement chez ceux atteints du sida avec une fréquence de 0,95 cas pour 1000 [8].
Lorsque les entités syndrome de Stevens-Johnson et syndrome de Lyell étaient séparées, on notait une prédilection pour l’homme (sex-ratio de 3:1) dans le premier, et pour la femme (sex-ratio de 1,5:1 à 2:1) dans le second. Le pic de fréquence de ces syndromes est compris entre 20 et 40 ans [6, 7]. Cependant ces affections ont aussi été rapportées chez des jeunes enfants et des vieillards. Il ne semble pas y avoir de prédilection pour une race ou une ethnie particulière [6, 7]. Une association statistiquement significative a été retrouvée dans certaines populations ethniques entre certains allèles HLA-B et le syndrome Stevens-Johnson/Lyell induit par l’allopurinol et la carbamazépine [9, 10]. Une recherche de l’allèle HLA-B*5701 doit aussi être systématiquement réalisée chez les patients devant recevoir de l’abacavir pour une infection au virus de l’immunodéficience humaine (VIH) [11].
L’incidence des complications oculaires est variable selon les séries mais on peut considérer que 70 à 80 % des patients hospitalisés pour ces syndromes sont ou seront concernés [12]. À l’inverse des lésions des autres muqueuses, les complications oculo-palpébrales n’existent pas uniquement lors de la phase aiguë cutanée de la maladie, elles peuvent aussi apparaître à distance de l’épisode initial [5].
Classiquement, le syndrome de Stevens-Johnson était considéré comme le résultat d’un conflit immunologique à complexes immuns circulants dans les parois vasculaires qui provoquait une vascularite « allergique ». Les facteurs déclenchants peuvent être notamment des agents infectieux, des médicaments, certaines tumeurs et quelques collagénoses [13]. Parmi les causes infectieuses, Mycoplasma pneumoniae et le virus Herpes simplex hominis sont le plus souvent cités [14]. Dans 50 à 60 % des syndromes de Stevens-Johnson, il existe à l’origine une prise de médicaments, au premier rang desquels figurent les sulfamides, certains anti-épileptiques (carbamazépine, phénytoïne, barbituriques), l’allopurinol et les anti-inflammatoires non stéroïdiens de type oxicam [6]. Même l’instillation de collyres, tels que la scopolamine, des sulfamides, la proparacaïne et le tropicamide (Mydriaticum®), a été incriminée [15]. Le syndrome de Lyell était plutôt attribué à une toxicité médicamenteuse directe idiosyncrasique. Cependant la cause déclenchante n’est souvent pas mise en évidence et le tableau clinique dans les formes sévères n’est évocateur d’aucun des deux syndromes. C’est pourquoi actuellement, dans un souci de simplification, la majorité des auteurs réunissent ces deux identités sous la même dénomination de syndrome de Stevens-Johnson/Lyell. Le mécanisme en cause est probablement immuno-allergique dans les deux cas.
Au niveau des muqueuses extra-oculaires et de la peau, les bulles sous-épidermiques contiennent des cellules monocytaires (histiocytes et lymphocytes). Il existe des infiltrats périvasculaires faits de cellules mononucléées. Lors de la phase aiguë de la maladie, il existe une déplétion importante en lymphocytes T auxiliaires ou helper (CD4) dont le taux redevient normal au moment de la guérison. Des études ont mis en évidence une augmentation des lymphocytes T suppresseurs (CD8) dans l’exsudat contenu dans les bulles cutanées. L’activation des lymphocytes T cytotoxiques CD8 et des macrophages, l’IL-6, le TNF-α, le ligand Fas (Fas-L) soluble et la granulysine seraient tous impliqués dans la pathogénie du syndrome de Stevens Johnson/Lyell [16].
L’analyse de conjonctives biopsiées lors de la phase aiguë montre une réaction inflammatoire non spécifique avec nécrose des artères et des veinules associée à une dégénérescence fibrinoïde du collagène. Cette inflammation se combine à une prolifération des cellules de la couche basale épithéliale. Lors de la phase chronique, une fibrose sous-épithéliale apparaît avec diminution très importante des cellules à mucus. Des complexes immuns composés d’immunoglobulines G, A et M, et des fractions C3 et C4 du complément peuvent être détectés dans la paroi des vaisseaux du plexus conjonctival, du chorion des muqueuses et des vaisseaux dermiques. Aucune de ces anomalies n’est cependant spécifique.
Contrairement aux traités classiques qui individualisent syndrome de Stevens-Johnson et syndrome de Lyell, ces deux entités seront réunies avec une différenciation en formes mineures et majeures. Classiquement, la dermatose peut apparaître jusqu’à 3 semaines après une première prise médicamenteuse. En cas de réexposition à la drogue inductrice, la réaction peut se développer en quelques heures. La durée de la phase aiguë de la maladie est en moyenne de 2 à 4 semaines [17].
Les diagnostics différentiels sont la dermatose à IgA linéaire, le pemphigus paranéoplasique, le pemphigus vulgaire, la pemphigoïde bulleuse, la pustulose exanthématique aiguë généralisée, l’érythème pigmenté fixe et l’épidermolyse aiguë staphylococcique ou maladie de Ritter von Rittershain (staphyloccocal scalded skin syndrome).
Les formes mineures correspondent à l’érythème polymorphe associé à une atteinte des muqueuses initialement décrit par Stevens et Johnson [2, 4]. Les prodromes de la maladie précèdent généralement les lésions cutanées. Cette phase initiale est constituée d’un malaise général souvent fébrile avec céphalées voire prostration. Dans les formes classiques, le diagnostic clinique est facile. Les lésions cutanées sont volontiers symétriques et plus fréquentes sur les faces d’extension des extrémités, surtout dos des mains et des pieds, mais aussi poignets, coudes et genoux. Le tronc n’est atteint que dans les formes les plus sévères. L’éruption est fugace, prenant l’aspect en certains endroits de cocarde érythémateuse évoluant rapidement vers un stade papuleux (fig. 7-8). Des vésicules et des bulles peuvent se développer, disparaissant alors sans laisser de cicatrice. Des lésions unguéales séquellaires sont par contre habituelles (fig. 7-9).
Les lésions des muqueuses extra-oculaires peuvent toucher les lèvres, les muqueuses de la bouche (fig. 7-10), du pharynx, du larynx, de la trachée, des bronches, de l’œsophage et les muqueuses génitales. Elles se manifestent sous la forme d’érosions polycycliques douloureuses, le stade vésiculobulleux étant trop fugace pour être visible. Elles disparaissent sans séquelle cicatricielle à l’inverse de l’atteinte conjonctivale qui fait à distance toute la gravité de la maladie, car elle est potentiellement cécitante.

Fig. 7-8 Lésions érythémateuses en cocarde sur les mains caractéristiques des formes mineures de syndrome de Stevens-Johnson/Lyell.
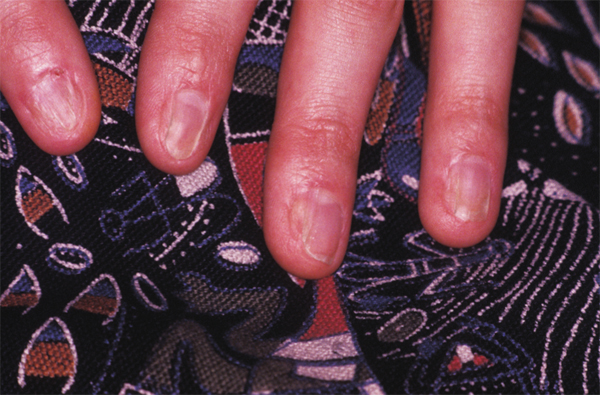
Fig. 7-9 Lésions unguéales séquellaires d’un syndrome de Stevens-Johnson/Lyell.
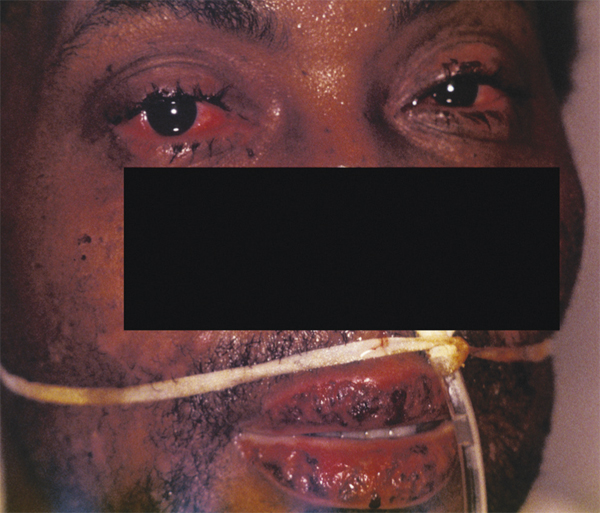
Fig. 7-10 Atteinte cutanéomuqueuse du visage au cours de la phase aiguë d’un syndrome de Stevens-Johnson/Lyell.
C’est au cours de la phase aiguë de ces formes sévères de la maladie que le pronostic vital peut être engagé, le taux de mortalité pouvant atteindre 30 %. Les principales complications sont la pneumonie, la septicémie, la myocardite, les myosites et la glomérulonéphrite. Le malaise général est souvent important et s’associe à une sensation de cuisson au niveau cutané et conjonctival. Initialement, l’éruption est morbilliforme et touche le visage et les extrémités. Elle s’étend ensuite au tronc et les lésions peuvent atteindre plus de 30 % de la surface corporelle en coalescent. Elles forment des bulles qui se décollent et laissent le derme « à nu ». La recherche du signe de Nikolsky est positive : le frottement de la peau en zone saine provoque un décollement épidermique. Lors de la phase aiguë, la muqueuse la plus fréquemment atteinte est la muqueuse buccale. Des infections graves, voire mortelles, peuvent compliquer cette phase aiguë. Une autre cause de morbidité est liée aux perturbations hydro-électrolytiques.
À long terme, il existe peu de séquelles cutanées. Une hyperpigmentation ou une hypopigmentation disparaissent en quelques mois. Les éventuelles cicatrices sont liées à une surinfection. Quelques cas d’alopécie définitive ont été rapportés.
Dans les formes mineures de la maladie, des complications oculaires surviennent entre 15 et 75 % des cas selon les études [12]. L’atteinte oculaire de la phase aiguë de la maladie est cliniquement très polymorphe. Il peut s’agir d’une inflammation palpébrale associant une dermatose de la face externe des paupières à une blépharite d’importance variable. Dans les formes sévères, la complication oculaire la plus fréquente est une conjonctivite non spécifique qui précède parfois l’éruption cutanée (fig. 7-11). Cette conjonctivite peut être catarrhale, pseudo-membraneuse ou purulente en cas de surinfection bactérienne. Dans quelques cas, on constate une érosion cornéenne persistante pendant 2 à 4 semaines. L’uvéite antérieure est exceptionnelle. Ces complications sont en général bilatérales. En fait, la morbidité oculaire du syndrome de Stevens-Johnson est liée aux séquelles cicatricielles conjonctivales.

Fig. 7-11 Syndrome de Stevens-Johnson/Lyell : conjonctivite à la phase aiguë.
Les complications oculaires chroniques de la maladie sont extrêmement graves, car elles sont souvent responsables de malvoyance et de cécité irréversibles. Que le degré de sévérité de l’atteinte oculaire à la phase aiguë représente un indicateur pronostic de complications oculaires tardives n’est pas toujours obligatoire [18, 19]. Celles-ci intéressent la conjonctive sous la forme d’une conjonctivite fibrosante rapidement progressive en rapport avec le processus inflammatoire, avec comblement des culs-de-sac conjonctivaux, formation de symblépharons (fig. 7-12) et d’un ankyloblépharon. La sécheresse oculaire, conséquence de ces remaniements conjonctivaux, est la complication tardive la plus fréquente [18]. Des réactivations inflammatoires conjonctivales sans récidives cutanées ont été attribuées à de possibles embols de complexes immuns circulants à herpèsvirus par exemple [5]. Des cas tardifs de sclérite diffuse et parfois nécrosante ont également été rapportés [20].
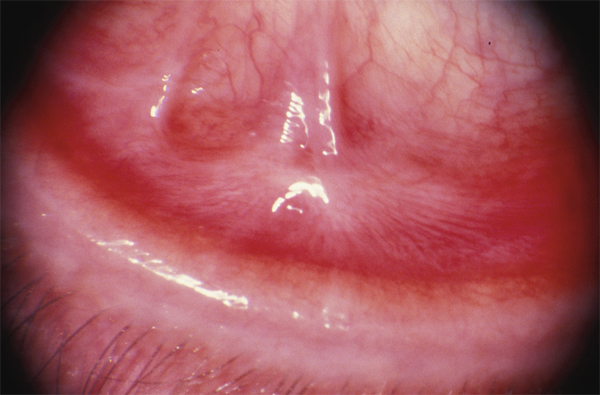
La cornée peut aussi perdre sa transparence par plusieurs mécanismes :
kératinisation cornéoconjonctivale secondaire (fig. 7-13) à la sécheresse oculaire et aboutissant au xérosis cornéen (fig. 7-14). Le film lacrymal est déficient quantitativement et qualitativement à cause de l’obstruction par la fibrose des orifices d’abouchement des glandes lacrymales, de la meibomite chronique associée et de la disparition des cellules à mucus conjonctivales ;
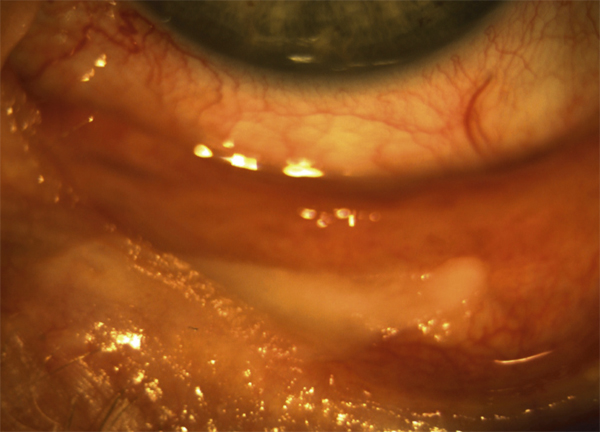
défects épithéliaux cornéens persistants dus à une irritation mécanique par des cils ectopiques, un entropion–trichiasis ou des bords libres palpébraux kératinisés ;
surinfections, opacités vascularisées et astigmatisme irrégulier de la cornée consécutifs.
Les patients sont généralement hospitalisés en unité de soins intensifs. Les médicaments soupçonnés d’avoir déclenché la maladie doivent être retirés. Toute nouvelle prise de ceux-ci est susceptible d’induire une récidive du syndrome mucocutané [21]. Les mesures thérapeutiques visent à compenser les pertes d’électrolytes et d’eau consécutives à l’altération de la fonction « barrière » de la peau. À la phase précoce, un score pronostique SCORTEN (score of toxic epidermal necrosis) détermine l’indice de gravité [19]. La prévention et le traitement d’une éventuelle surinfection, qui est responsable de la moitié de la mortalité lors de la phase aiguë, sont primordiaux [22]. La corticothérapie générale par voie systémique est discutée [12, 23]. Certaines études tendent à démontrer que celle-ci, administrée à forte dose, serait dans certains cas bénéfique sur l’intensité de la nécrose épithéliale et sur le pronostic oculaire, en association avec une corticothérapie locale [24]. Malheureusement, elle favorise les surinfections et n’est pas dénuée d’autres effets secondaires. Des perfusions intraveineuses précoces d’importantes quantités d’immunoglobulines G (doses cumulatives moyennes de 1,5 à 3,5 g/kg) permettraient pour certains d’améliorer le pronostic vital, avec dans l’ensemble une bonne tolérance [25], mais pour d’autres elles sont inefficaces [26]. Aucun autre traitement n’a sinon fait preuve d’une réelle efficacité, que ce soit le cyclophosphamide, la ciclosporine ou les plasmaphérèses.
Le traitement oculaire lors de la phase aiguë repose essentiellement sur une lubrification efficace des yeux au moyen de sérum physiologique, de gels dépourvus d’agents conservateurs et de pommade ophtalmique à la vitamine A. Les collyres antibiotiques ne sont utilisés qu’en cas de surinfection et les cycloplégiques qu’en cas d’uvéite antérieure aiguë. Associés aux corticoïdes par voie générale, les corticoïdes locaux seraient bénéfiques aux yeux [24]. Cependant, ils facilitent les surinfections conjonctivales et cornéennes chez ces patients dont l’épithélium de la surface oculaire est endommagé. Leur efficacité en monothérapie locale n’est pas prouvée dans la prévention de la formation des symblépharons au décours de la phase aiguë, pas plus d’ailleurs que celle des manœuvres instrumentales visant à préserver les culs-de-sac conjonctivaux (conformateurs, libération mécanique régulière des adhérences). Les corticoïdes locaux peuvent par contre être utilisés de manière discontinue pour limiter la gêne fonctionnelle occasionnée par l’inflammation. Le rôle de la ciclosporine topique n’a pas encore été étudié.
Le traitement médical des complications oculaires chroniques a pour principal objectif de soulager le syndrome de sécheresse oculaire quasi constant et de guérir les surinfections locales. Il est en cela identique à celui indiqué pour la phase aiguë de la maladie. L’acide transrétinoïque topique pourrait diminuer la transdifférenciation épithéliale consécutive à l’agression de l’épithélium conjonctival, mais il n’est pas commercialisé [27]. Les mucolytiques locaux et généraux, comme la N-acétyl-cystéine, diminueraient la formation de filaments, mais leur efficacité est loin d’être démontrée. Le bévacizumab en collyre a permis de faire régresser des néovaisseaux cornéens séquellaires [28]. En cas de persistance d’une inflammation sévère ou d’une récidive inflammatoire résistant aux anti-inflammatoires locaux, on peut recourir à une immunosuppression systémique au moyen d’une corticothérapie générale et/ou d’immunosuppresseurs cytotoxiques comme l’azathioprine (Imurel®) ou le cyclophosphamide (Endoxan®) [29]. Les règles d’utilisation de ces traitements sont alors similaires à celles régissant le traitement des maladies bulleuses auto-immunes. Un traitement par anticorps monoclonaux par voie systémique peut être tenté dans les formes réfractaires [30].
Au cours de la phase aiguë de la maladie, une greffe de membrane amniotique peut s’avérer très utile, parfois associée à une greffe cornéenne lamellaire ou transfixiante à chaud pour une perforation cornéenne [31]. Au décours de la phase aiguë, quand les lésions sont stabilisées ou pendant la phase d’inflammation chronique, la chirurgie conjonctivopalpébrale reconstructrice [32] et de restitution de la transparence cornéenne répond aux mêmes règles que celles établies pour les conjonctivites auto-immunes fibrosantes. Les techniques de reconstruction de la surface oculaire font appel de manière diversement associée : aux greffes de cornée lamellaires ou transfixiantes ; aux greffes de cellules souches limbiques en général allogéniques et non autologues à cause de la bilatéralité habituelle de l’atteinte oculaire ; aux allogreffes de cellules souches limbiques ou aux cellules souches limbiques cultivées in vitro ; aux greffes de membrane amniotique et enfin, plus récemment, aux greffes de muqueuse buccale transdifférenciée in vitro en épithélium cornéen [33–37]. En cas d’échec et pour les formes très sévères de déficience en cellules souches limbiques avec xérosis, il reste encore la possibilité de proposer une kératoprothèse [38] ou une ostéo-odontoprothèse [39], mais dont le pronostic à long terme reste très réservé.
[1] Hebra F. On diseases of the skin, including the exanthematha. vol. 1. Translated and edited by CH Fagge. London : New Sydenham Society ; 1866.
[2] Stevens AM, Johnson FC. A new eruptive fever associated with stomatitis and ophthalmia : report of two cases in children. Am J Dis Child 1922 ; 24 : 526-33.
[3] Lyell A. Toxic epidermal necrolysis : an eruption resembling scalding of the skin. Br J Dermatol 1956 ; 68 : 355-61.
[4] Bastuji-Garin S, Rzany B, Stern RS. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol 1993 ; 129 : 92-6.
[5] Foster CS, Fong LP, Azar D, et al. Episodic conjunctival inflammation after Stevens-Johnson syndrome. Ophthalmology 1988 ; 95 : 453-62.
[6] Chan HL, Stern RS, Arndt KA. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis : a population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol 1990 ; 126 : 43-7.
[7] Roujeau JC, Guillaume JC, Fabre JP. Toxic epidermal necrolysis (Lyell syndrome) : incidence and drug etiology in France, 1981-1985. Arch Dermatol 1990 ; 126 : 37-42.
[8] Fischel MA, Dickinson GM. Fansidar prophylaxis of pneumocystis pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986 ; 10 : 629.
[9] Cheung YK, Cheng SH, Chan EJ, et al. HLA-B alleles associated with severe cutaneous reactions to antiepileptic drugs in Han Chinese. Epilepsia 2013 ; 54 : 1307-14.
[10] Somkrua R, Eickman EE, Saokaew S, et al. Association of HLA-B*5801 allele and allopurinol-induced Stevens Johnson syndrome and toxic epidermal necrolysis : a systematic review and meta-analysis. BMC Med Genet 2011 ; 12 : 118.
[11] Allal S, Phillips E, Carosi G. HLA-B*5701 screening for hypersensitivity to Abacavir. N Eng J Med 2008 ; 358 : 568-79.
[12] Power WJ, Ghoraishi M, Merayo-Lloves J, et al. Analysis of the acute ophthalmic manifestations of the erythema multiforme/Stevens-Johnson syndrome/toxic epidermal necrolysis disease spectrum. Ophthalmology 1995 ; 102 : 1669-76.
[13] Wuepper KD, Watson PA, Kazmierowski JA. Immune complexes in erythema multiforme and Stevens-Johnson syndrome. J Invest Dermatol 1980 ; 74 : 368-71.
[14] Kim PS, Goldfarb IW, Gaisford JC, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis : a pathophysiologic review with recommendations for a treatment protocol. J Burn Care Rehabil 1983 ; 4 : 91-100.
[15] Genvert GI, Cohen EJ, Donnenfeld ED, et al. Erythema multiforme after use of topical sulfacetamide. Am J Ophthalmol 1985 ; 99 : 465-8.
[16] Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis : review of pathogenesis and management. J Am Acad Dermatol 2012 ; 66 : 995-1003.
[17] Revuz J, Penso D, Roujeau JC, et al. Toxic epidermal necrolysis : clinical findings and prognosis factors in 87 patients. Arch Dermatol 1987 ; 123 : 1160-5.
[18] Gueudry J, Roujeau JC, Binaghi M, et al. Risk factors for the development of ocular complications of Stevens-Johnson syndrome and toxic epidermal necrolysis. Arch Dermatol 2009 ; 145 : 157-62.
[19] Morales ME, Purdue GF, Verity SM, et al. Ophthalmic manifestations of Stevens-Johnson syndrome and toxic epidermal necrolysis and relation to SCORTEN. Am J Ophthalmol 2010 ; 150 : 505-10.
[20] Robin H, Hoang-Xuan T, Bouder P, et al. Le syndrome de Stevens-Johnson, une nouvelle cause de sclérite ? Bull Soc Ophtalmol Fr 1993 ; 9 : 665-72.
[21] Patterson R, Dykewicz MS, Gonzales A. Erythema multiforme and Stevens-Johnson syndrome. Descriptive and therapeutic controversy. Chest 1990 ; 98 : 331-6.
[22] Revuz J, Roujeau JC, Guillaume JC. Treatment of toxic epidermal necrolysis : Créteil’s experience. Arch Dermatol 1987 ; 123 : 1156-8.
[23] Halebian PH, Madden MR, Finklestein JL. Improved burn center survival of patients with toxic epidermal necrolysis managed without corticosteroids. Ann Surg 1986 ; 204 : 503-12.
[24] Araki Y, Sotozono C, Inatomi T, et al. Successful treatment of Stevens-Johnson syndrome with steroid pulse therapy at disease onset. Am J Ophthalmol 2009 ; 147 : 1004-11.
[25] French LE, Trent JT, Kerdel FA. Use of intravenous immunoglobulin in toxic epidermal necrolysis and Stevens-Johnson syndrome : our current understanding. Int Immunopharmacol 2006 ; 6 : 543-9.
[26] Yip LW, Thong BY, Tan AW, et al. High-dose intravenous immunoglobulin in the treatment of toxic epidermal necrolysis : a study of ocular benefits. Eye 2005 ; 19 : 846-53.
[27] Soong HK, Martin NF, Wagoner MD. Topical retinoid therapy for squamous metaplasia of various ocular surface disorders : a multicenter, placebo-controlled double-masked study. Ophthalmology 1988 ; 95 : 1442-6.
[28] Uy HS, Chan PS, Ang RE. Topical bevacizumab and ocular surface neovascularization in patients with Stevens-Johnson syndrome. Cornea 2008 ; 27 : 70-3.
[29] Henz MC, Allen SG. Efficacy of cyclophosphamide in toxic epidermal necrolysis : clinical and pathophysiologic aspects. J Am Acad Dermatol 1991 ; 25 : 778-86.
[30] Fiorelli VM, Dantas PE, Jackson AT, et al. Systemic monoclonal antibody therapy (daclizumab) in the treatment of cicatrizing conjunctivitis in Stevens-Johnson syndrome, refractory to conventional therapy. Curr Eye Res 2010 ; 35 : 1057-62.
[31] Shay E, Kheirkhah A, Liang L, et al. Amniotic membrane transplantation as a new therapy for the acute ocular manifestations of Stevens-Johnson syndrome and toxic epidermal necrolysis. Surv Ophthalmol 2009 ; 54 : 686-96.
[32] Kheirkhah A, Ghaffari R, Kaghazkanani R, et al. A combined approach of amniotic membrane and oral mucosa transplantation for fornix reconstruction in severe symblepharon. Cornea 2013 ; 32 : 155-60.
[33] Tseng SCG, Prabhasawat P, Barton K, et al. Amniotic membrane transplantation with or without limbal allografts for corneal surface reconstruction in patients with limbal stem cell deficiency. Arch Ophthalmol 1998 ; 116 : 431-41.
[34] Pellegrini G, Traverso CE, Franzi AT, et al. Long-term restoration of damaged corneal surfaces with autologous cultivated corneal epithelium. Lancet 1997 ; 349 : 990-3.
[35] Omoto M, Shimmura S, Hatou S, et al. Simultaneous deep anterior lamellar keratoplasty and limbal allograft in bilateral limbal stem cell deficiency. Jpn J Ophthalmol 2010 ; 54 : 537-43.
[36] Satake Y, Higa K, Tsubota K, et al. Long-term outcome of cultivated oral mucosal epithelial sheet transplantation in treatment of total limbal stem cell deficiency. Ophthalmology 2011 ; 118 : 1524-30.
[37] Sotozono C, Inatomi T, Nakamura T, et al. Visual improvement after cultivated oral mucosal epithelial transplantation. Ophthalmology 2013 ; 120 : 193-200.
[38] Pujari S, Siddique SS, Dohlman CH, et al. The Boston keratoprosthesis type II : the Massachusetts eye and ear infirmary experience. Cornea 2011 ; 30 : 1298-303.
[39] Tan A, Tan DT, Tan XW, et al. Osteo-odonto keratoprosthesis : systematic review of surgical outcomes and complication rates. Ocul Surf 2012 ; 10 : 15-25.
T. Hoang-Xuan
La maladie du greffon contre l’hôte résulte d’un conflit immunitaire provoqué par des cellules immunocompétentes résiduelles provenant des cellules souches hématopoïétiques allogéniques du donneur. La complication oculaire essentielle est la sécheresse oculaire survenant au stade chronique de la maladie. Elle peut perturber très sévèrement le confort oculaire et la qualité de vie du patient, et être responsable de complications cornéennes parfois cécitantes. Le traitement de cette sécheresse oculaire est souvent très décevant.
La maladie du greffon contre l’hôte (MGCH) dans sa forme chronique est la première cause de morbidité à long terme et de mortalité après transplantation allogénique de cellules souches hématopoïétiques. Celle-ci, appelée plus communément greffe de moelle, représente la solution thérapeutique ultime pour certaines hémopathies malignes, aplasies médullaires et maladies héréditaires de surcharge.
Les complications oculaires de la MGCH chronique sont fréquentes et parfois graves. Elles sont redoutées, car elles altèrent très souvent et significativement la qualité de vie des patients.
Pour que la MGCH puisse se déclarer, très schématiquement, trois conditions sont nécessaires : la greffe contient des cellules immunocompétentes ; l’hôte possède de nombreux iso-antigènes manquant chez le donneur ; l’hôte ne peut développer une réponse immune efficace. La pathophysiologie de la maladie est en fait bien plus complexe avec de nombreuses inconnues. Elle fait intervenir des mécanismes immunitaires impliquant les lymphocytes B et T du donneur et de nombreuses autres cellules dans un réseau d’interactions compliquées [1]. Une communauté d’identité HLA entre patient et donneur n’empêche pas toujours la maladie de se déclarer à cause de différences génétiques en dehors des loci HLA ou d’antigènes d’histocompatibilité mineurs.
L’incidence de la MGCH est élevée, la maladie survenant avec un degré variable dans la moitié des cas après transplantation de cellules souches hématopoïétiques. Le pronostic de la MGCH est très sévère, de nombreux patients décédant soit de complications de la maladie, soit des effets secondaires de la chimiothérapie immunosuppressive.
Par le passé, on considérait la maladie comme aiguë avant son centième jour d’évolution et comme chronique après. En 1975, une conférence de consensus du National Institutes of Health (NIH) a essayé de redéfinir la classification de la maladie qui comporte toujours deux phases, aiguë et chronique, mais avec au sein de chacune deux sous-catégories, la notion du centième jour disparaissant [2]. L’incidence de la forme chronique est d’environ 50 %.
La phase aiguë comporte une forme classique, survenant 20 jours après la greffe, et caractérisée par une éruption cutanée papulomaculaire érythémateuse accompagnée d’un prurit et d’une sensation de coup de soleil. Elle intéresse la paume des mains, la plante des pieds, les oreilles, puis le tronc, le visage et les extrémités. Une évolution vers des bulles laissant place à des lésions érosives est aussi possible. Les atteintes viscérales intéressent essentiellement les intestins sous la forme de diarrhées douloureuses et le foie avec une hépatite cholestatique.
Selon la conférence de consensus du NIH, il existe une autre sous-catégorie de MGCH aiguë pouvant apparaître au-delà du centième jour de la maladie, soit dans la continuité du tableau classique, soit sous la forme d’une récidive, soit de novo [2].
Les complications de la MGCH aiguë sont les hémorragies, les infections (bactériennes, mycosiques, herpétiques dont la pneumopathie interstitielle à cytomégalovirus) et les effets secondaires de la chimiothérapie.
On a longtemps considéré l’existence d’une seule MGCH chronique qui survient à partir du centième jour après la greffe. Elle se manifeste après greffe de moelle allogénique, soit après une phase de rémission de la maladie aiguë, soit dans la continuité de la phase aiguë mal contrôlée. Il existe un second sous-groupe de forme chronique, beaucoup plus rare, dans laquelle on retrouve des manifestations caractéristiques de la forme aiguë, et qui peut aussi survenir de novo [2].
Le diagnostic de la MGCH est en général facile cliniquement. Les manifestations cliniques les plus caractéristiques de la MGCH chronique sont l’atteinte cutanée sclérodermiforme ou lichénoïde, la poïkilodermie (fig. 7-15), le lichen plan buccal et génital, les plaques kératosiques de la bouche, les fibroses et sténoses vaginales, le reflux et la sténose de l’œsophage, les fasciites et les contractures articulaires, et la bronchiolite obstructive à la biopsie pulmonaire. La dépigmentation, la dystrophie unguéale, l’alopécie, la xérostomie, les ulcères génitaux et les myosites sont d’autres signes évocateurs mais non contributifs, à eux seuls, au diagnostic. La présentation de la maladie peut être très polymorphe avec une atteinte tissulaire isolée ou multisystémique. La biopsie peut parfois aider à distinguer les formes frontières entre MGCH aiguë et chronique. Des biomarqueurs de la phase chronique ont été aussi mis en évidence récemment. Parmi les complications tardives figurent les infections dont notamment celles à Pneumocystis carinii et le zona, et les cancers secondaires comme les carcinomes épidermoïdes. La mortalité à 10 ans après le début de la MGCH chronique est de 60 %.
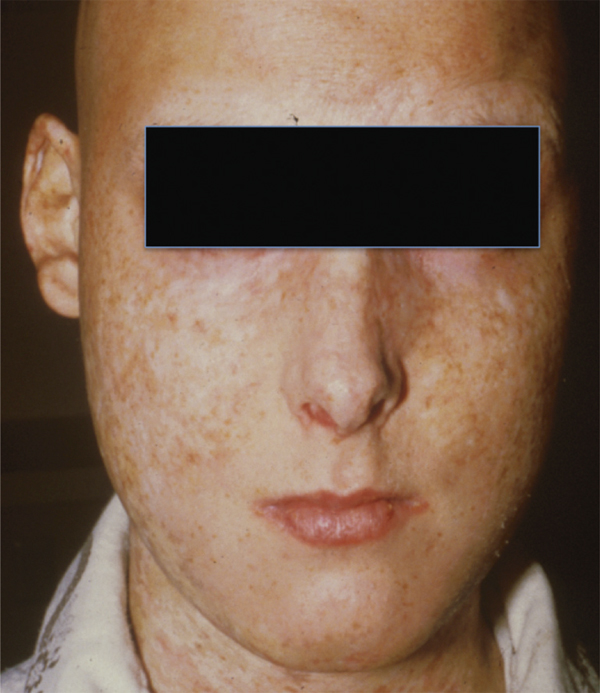
Fig. 7-15 Poïkilodermie et calvitie.
La complication oculaire qui caractérise la MGCH aiguë est la conjonctivite. Elle survient 2 semaines après le début de la maladie. Elle serait de mauvais pronostic avec 90 % de mortalité si elle s’accompagne de chémosis, d’exsudation sérosanguine, de pseudo-membranes et/ou de désépithélialisation cornéenne. Douze pour cent des patients avec MGCH aiguë développent une conjonctivite de ce type [3].
Les signes oculaires au cours de la MGCH chronique sont en rapport avec l’atteinte de la conjonctive et de la glande lacrymale. Onze pour cent des patients développent une conjonctivite chronique qui peut se compliquer de fibrose comme dans la pemphigoïde cicatricielle [3]. La conjonctivite s’accompagne souvent d’une atteinte systémique. Des donneurs allergiques peuvent aussi transmettre des allergies oculaires [4].
Des lésions palpébrales sont possibles à type de poliose, madarose, lagophtalmie et entropion.
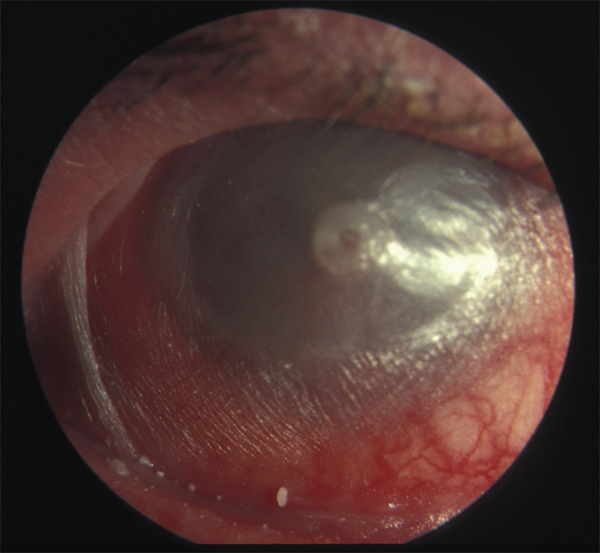
L’atteinte inflammatoire des glandes lacrymales survient au cours de la MGCH aiguë et chronique. Elle est responsable d’une sécheresse oculaire quasi irréversible qui est aggravée par une éventuelle fibrose conjonctivale. Sa survenue précoce est un facteur de mauvais pronostic visuel car la sécheresse oculaire est souvent majeure (fig. 7-16) et les complications cornéennes inévitables et sévères (fig. 7-17 et 7-18) : kératite filamenteuse, kératopathie superficielle ponctuée, ulcères cornéens, fonte cornéenne aboutissant à la perforation. Environ la moitié des patients avec MGCH chronique développent une sécheresse oculaire similaire à celle du syndrome de Gougerot-Sjögren. Des sclérites antérieures et postérieures ont aussi été décrites [5].
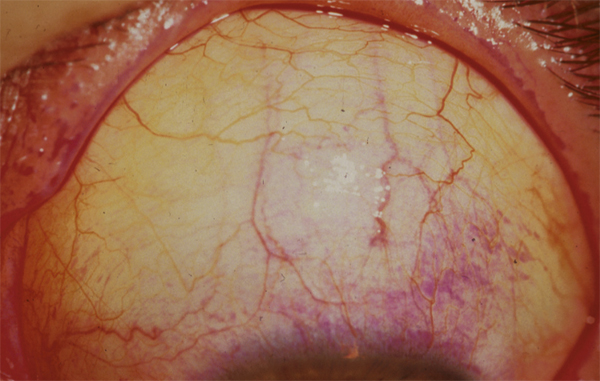
Fig. 7-16 Sécheresse oculaire : la conjonctive est marquée par le rose Bengale.
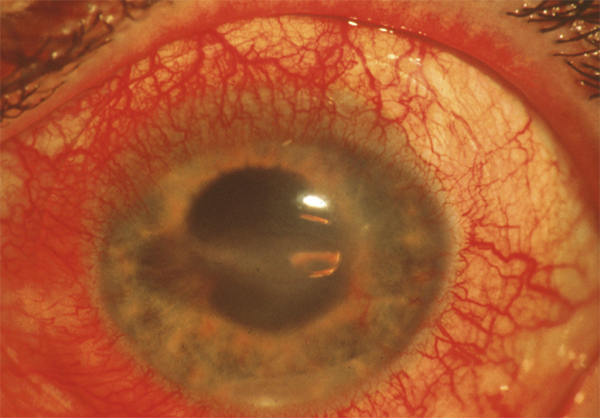
Fig. 7-17 Ulcération cornéenne trophique dans l’aire de la fente interpalpébrale par sécheresse oculaire.

Fig. 7-18 Atteinte cornéenne très sévère avec taie diffuse néovascularisée.
Cataracte : la corticothérapie systémique et l’irradiation corporelle sont responsables de cataracte à 5 ans chez environ 25 % des patients souffrant de MGCH chronique.
Infections oculaires : l’immunodépression médicamenteuse induite est source d’endophtalmie fongique en particulier à Candida albicans dans 1,5 à 4 % des cas, rarement dans la période immédiate suivant la greffe. Tardivement et dans moins de 0,5 % des cas peuvent survenir une rétinite virale à Herpes zoster virus ou à cytomegalovirus et une rétinite toxoplasmique.
Rétinopathies : la prévalence de la microvasculopathie occlusive chez les leucémiques greffés de moelle est de 4 à 10 %. Elle survient au cours de la MGCH chronique et les causes incriminées sont la ciclosporine, la chimiothérapie et l’irradiation corporelle totale. Les conséquences visuelles sont variables.
Autres complications : les hémorragies intrarétiniennes, vitréennes et sous-conjonctivales ; une récidive de rétinopathie leucémique ; un œdème papillaire bilatéral réversible sans baisse de la vision et induit par la ciclosporine (2 %) ; un décollement séreux du neuro-épithélium par coagulation intravasculaire disséminée (0,5 %).
Le traitement prophylactique de la MGCH aiguë comprend principalement les inhibiteurs de la calcineurine dont la ciclosporine et l’élimination des lymphocytes de la moelle du donneur par séparation mécanique ou sérum antilymphocytaire.
Le traitement curatif de la MGCH aiguë repose sur la prednisone et la ciclosporine dont il faut augmenter les doses dans les formes résistantes. Les associations avec l’azathioprine, le mycophénolate mofétil et la thalidomide ont été essayées avec des résultats mitigés.
Le traitement préventif de la MGCH chronique n’existe quasiment pas. Seules les globulines antithymocytes diminueraient l’incidence et la sévérité de la maladie [6]. Le rituximab semble aussi prometteur mais il manque encore de recul pour juger de sa réelle efficacité [7]. De nombreux progrès ont été néanmoins réalisés dans la prise en charge globale des patients pour les soulager de leur symptomatologie et prévenir les complications infectieuses plus particulièrement.
Le traitement curatif de la MGCH chronique est décevant et repose sur la corticothérapie par voie orale dont la posologie peut être diminuée grâce à la ciclosporine.
Localement, l’inflammation oculaire peut être soulagée par une corticothérapie locale dont il faut surveiller les effets secondaires potentiels [8]. La ciclosporine semble aussi apporter quelques bénéfices [9].
La sécheresse oculaire qui est la complication oculaire principale de la MGCH chronique n’est pas soulagée par les collyres anti-inflammatoires. Son traitement symptomatique repose sur les substituts des larmes, éventuellement le sérum autologue voire allogénique [10] et les bouchons lacrymaux. Une meibomite peut contribuer à l’œil sec par excès d’évaporation des larmes et doit être traitée par des mesures d’hygiène des paupières avec compresses chaudes et massages, voire par antibiothérapie par voie orale avec des cyclines ou ses dérivés. Dans les formes avec complications cornéennes sévères, on peut avoir recours à la greffe de membrane amniotique, aux différentes variantes de greffes de cellules souches limbiques, au recouvrement conjonctival, à la tarsorraphie et à la greffe de cornée. Récemment, les nouveaux verres scléraux permettent de prévenir ces complications cornéennes tout en améliorant significativement le confort oculaire et la vision [11].
[1] Socié G, Ritz J. Current issues in chronic graft-versus-host disease. Blood 2014 ; 124 : 374-84.
[2] Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease : I. diagnosis and staging working group report. Biol Blood Marrow Transplant 2005 ; 11 : 945-56.
[3] Jabs DA, Wingard J, Green WR, et al. The eye in bone marrow transplantation. III. Conjunctival graft-vs-host disease. Arch Ophthalmol 1989 ; 107 : 1343-8.
[4] Tabbara KF, Nassr A, Ahmed SO, et al. Acquisition of vernal and atopic keratoconjunctivitis after bone marrow transplantation. Am J Ophthalmol 2008 ; 146 : 462-5.
[5] Nassar A, Tabbara KF, Aljurf M. Ocular manifestations of graft-versus-host disease. Saudi J Ophthalmol 2013 ; 27 : 215-22.
[6] Socie G, Schmoor C, Bethge WA, et al. Chronic graft-versus-host disease : long-term results from a randomized trial on graft-versus-host disease prophylaxis with or without anti-t-cell globulin ATG-Fresenius. Blood 2011 ; 117 : 6375-82.
[7] Cutler C, Kim HT, Bindra B, et al. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation : results of a phase 2 trial. Blood 2013 ; 122 : 1510-7.
[8] Robinson MR, Lee SS, Rubin BI, et al. Topical corticosteroid therapy for cicatricial conjunctivitis associated with chronic graft-versus-host disease. Bone Marrow Transplant 2004 ; 33 : 1031-5.
[9] Wang Y, Ogawa Y, Dogru M, et al. Ocular surface and tear functions after topical cyclosporine treatment in dry eye patients with chronic graft-versus-host disease. Bone Marrow Transplant 2008 ; 41 : 293-302.
[10] Chiang CC, Lin JM, Chen WL, et al. Allogeneic serum eye drops for the treatment of severe dry eye in patients with chronic graft-versus-host disease. Cornea 2007 ; 26 : 861-3.
[11] Jacobs DS, Rosenthal P. Boston scleral lens prosthetic device for treatment of severe dry eye in chronic graft-versus-host disease. Cornea 2007 ; 26 : 1195-9.
E. Gabison, T. Hoang-Xuan
Les épisclérites sont des inflammations oculaires le plus souvent bénignes. Rarement, elles sont l’expression ophtalmologique de vascularites systémiques. Les sclérites sont des inflammations oculaires le plus souvent associées à des pathologies générales qui peuvent également impliquer la cornée, l’uvée, la rétine et le nerf optique et grever ainsi le pronostic visuel. Un interrogatoire et des examens biologiques ciblant les maladies systémiques les plus fréquemment associées sont nécessaires afin de dépister une vascularite potentiellement mortelle. Le traitement de ces affections nécessite généralement l’utilisation d’anti-inflammatoires généraux et/ou d’immunosuppresseurs dans le cadre d’une collaboration étroite entre ophtalmologistes et internistes et/ou rhumatologistes.
La sclère est la tunique blanche externe rigide de l’œil sur laquelle s’insèrent les muscles oculo-moteurs. Elle se prolonge en avant vers l’éperon scléral dans lequel est enchâssée la cornée et en arrière vers son foramen postérieur qui entoure le nerf optique. La sclère est constituée histologiquement de trois couches. L’épisclère est la plus superficielle, composée de fibres de collagène et de protéoglycanes peu compactées associées à des fibroblastes. Le stroma correspond à la couche intermédiaire. Il est constitué d’une matrice extracellulaire plus dense, pauvre en cellules (les sclérocytes), mais riche en collagène, élastine et protéoglycanes entrelacés qui rendent la sclère blanche opaque à l’état physiologique [1]. La lamina fusca est la couche la plus profonde en contact avec l’uvée. En cas d’inflammation, l’amincissement ou la simple désorganisation des fibres de la matrice sclérale peut rendre le stroma translucide et laisser deviner la pigmentation choroïdienne sous-jacente.
La sclère est nourrie par imbibition à partir de ses vascularisations superficielles (le plexus épiscléral profond) et profondes (la choroïde), ce qui la rend particulièrement sensible à l’ischémie. Sa frêle vascularisation s’oppose à son innervation riche (issue des nerfs ciliaires courts postérieurs en arrière et des nerfs ciliaires longs postérieurs en avant) qui donne aux sclérites leur caractère particulièrement douloureux.
Les inflammations sclérales peuvent être bénignes et autolimitées, ou sévères et nécessiter des thérapeutiques immunosuppressives. La topographie de l’inflammation et le type de plexus vasculaire impliqués permettent de distinguer les épisclérites, qui correspondent à des inflammations aiguës le plus souvent bénignes du plexus épiscléral superficiel, des sclérites plus chroniques, douloureuses et sévères qui intéressent le plexus épiscléral profond. Les épisclérites ont le plus souvent une étiologie bénigne, au contraire des sclérites qui peuvent révéler ou compliquer une pathologie systémique potentiellement cécitante et/ou mortelle [2].
La classification clinique de Watson et Hayreh sert toujours de référence pour établir le diagnostic et le pronostic des inflammations sclérales [3]. Elle aide à distinguer les différentes formes cliniques de sclérites et d’épisclérites et permet d’évaluer leur sévérité non seulement locale, mais aussi générale grâce à une revue exhaustive des associations possibles à des pathologies de système.
Avant tout, la rougeur oculaire doit être analysée à l’œil nu, idéalement à la lumière du jour. Cela permet d’identifier plus facilement les zones de modification de la coloration sclérale témoignant de poussées inflammatoires anciennes. La rougeur est aussi mieux appréciée, vive dans les épisclérites où c’est le plexus épiscléral superficiel qui est dilaté, plus sombre voire violacée dans les sclérites où le plexus épiscléral profond est surtout concerné. L’examen à la lampe à fente précise l’orientation des plexus épiscléraux, radiaire par rapport au limbe pour le superficiel et moins systématisé, transverse et entrecroisé pour le profond (fig. 7-19). Le test de vasoconstriction à la phényléphrine 10 % fait disparaître en quelques secondes la rougeur de l’épisclérite alors que celle de la sclérite persiste (fig. 7-20). En cas de vascularite ischémique avec nécrose sclérale, la rougeur peut laisser place à des zones blanches avasculaires à travers lesquelles on perçoit parfois la couleur sombre de la choroïde [4]. Des anomalies plus discrètes de la vascularisation sclérale, en particulier les zones d’ischémie, sont détectées plus aisément par l’examen biomicroscopique en lumière verte avec le filtre anérythre (fig. 7-21).
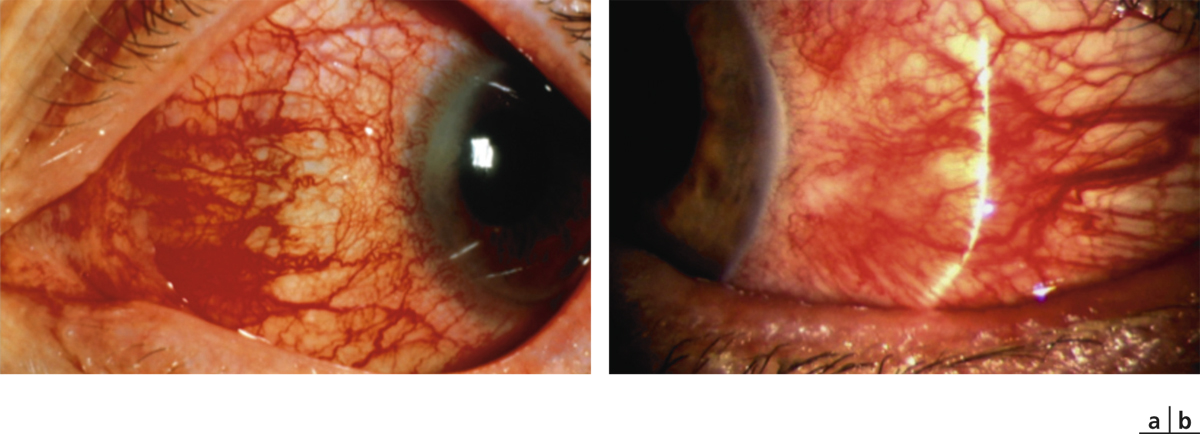
Fig. 7-19 Inflammation épisclérale et sclérale.
a. Épisclérite : noter l’orientation radiaire des vaisseaux par rapport au limbe.
b. Sclérite : noter l’aspect peigné oblique des vaisseaux par rapport au limbe.
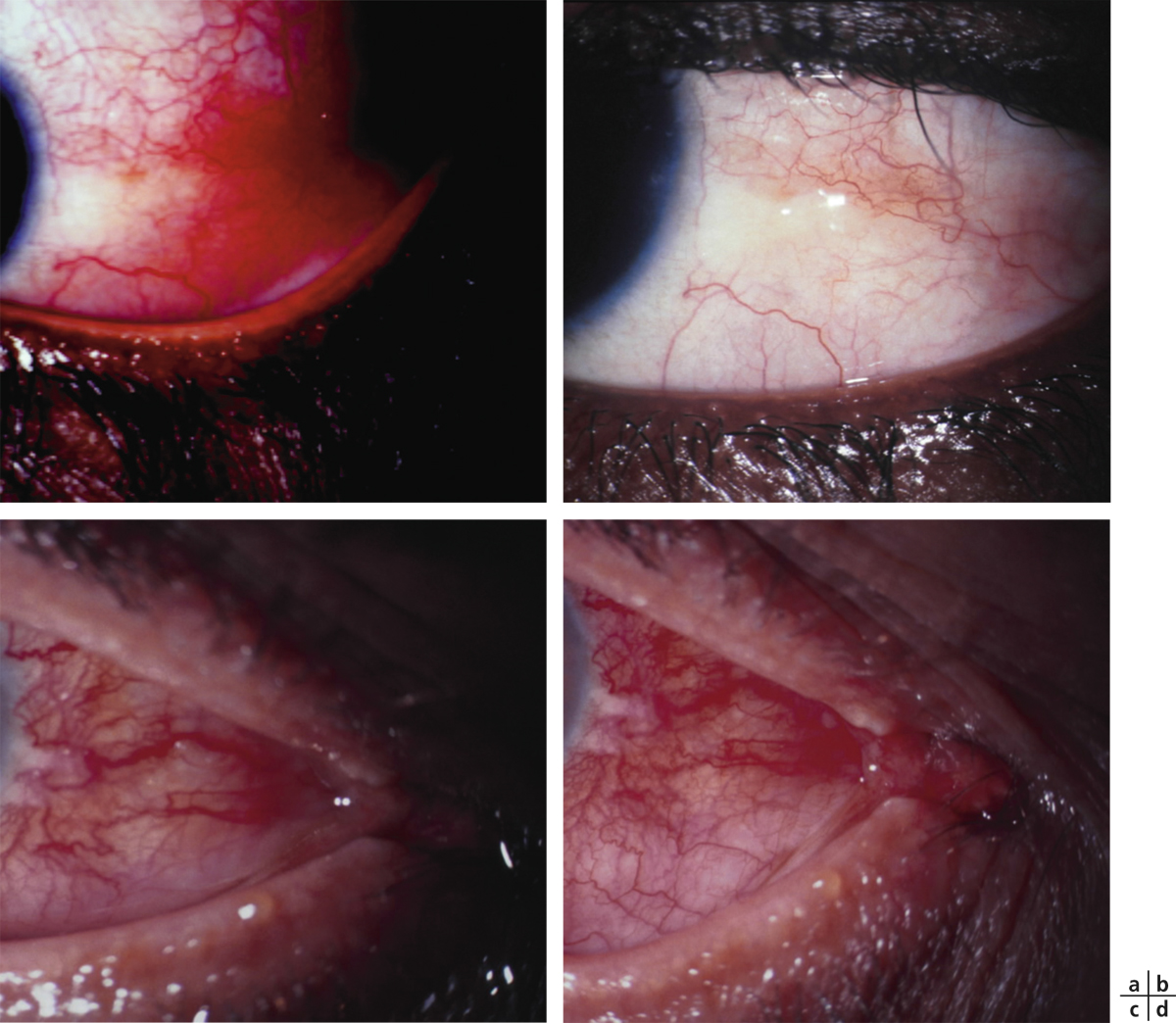
Test de vasoconstriction à la phényléphrine 10 % dans les inflammations épisclérales et sclérales (avant et après instillation).
a, b. Épisclérite : avant phényléphrine (a) ; après phényléphrine et noter la disparition de la rougeur en moins de 15 secondes (b).
c, d. Sclérite : avant phényléphrine (c) ; après phényléphrine noter la persistance de la rougeur (d).
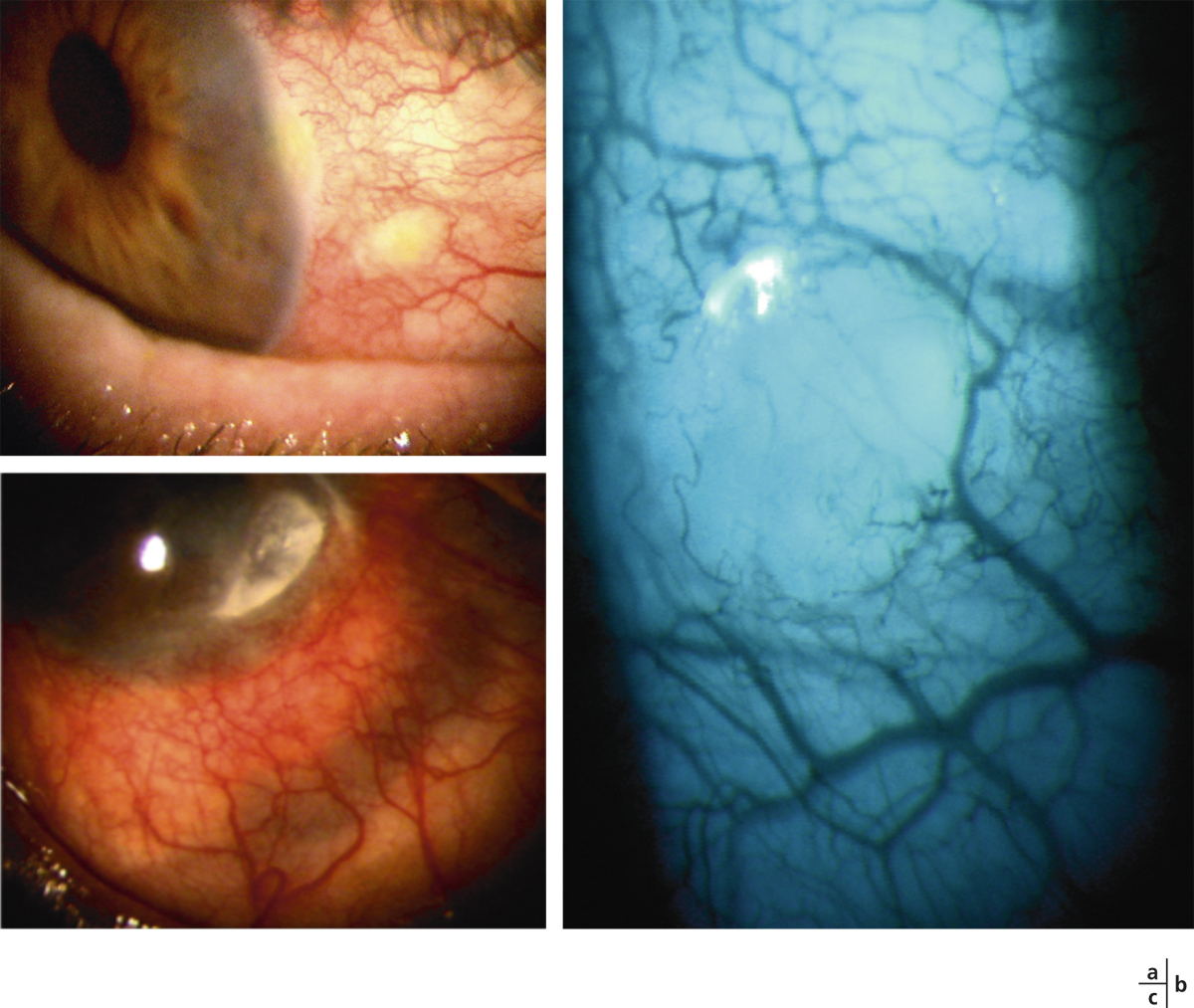
Fig. 7-21 Examen de la sclère en lumière diffuse.
a. La zone ovalaire d’ischémie sclérale sur le méridien de 3 heures est discrète.
b. Elle est mieux objectivée par l’analyse en lumière verte anérythre.
c. Modification de la coloration sclérale d’aspect plus sombre (séquelle de sclérite).
L’examen au biomicroscope en fente fine permet dans les cas difficiles de distinguer les inflammations sclérales des inflammations épisclérales. Il repère les deux lignes de profil correspondant à la sclère en profondeur et la conjonctive plus superficielle. Dans l’épisclérite, ces deux lignes sont exagérément séparées, comme dans un chémosis, alors que dans les sclérites, l’œdème scléral repousse la conjonctive et les lignes de profil sont parallèles, refoulées vers l’avant et accolées (fig. 7-22).

Lignes de profil dans les inflammations épisclérales et sclérales.
a. Épisclérite : noter la séparation des lignes de profil antérieure et postérieure.
b. Sclérite : noter que les lignes de profil antérieure et postérieure sont parallèles et accolées.
L’examen de la surface oculaire est particulièrement important, car l’épisclérite peut compliquer toutes les pathologies inflammatoires de la surface oculaire. Les sclérites, dont les étiologies sont à rechercher dans les maladies systémiques, peuvent aussi être provoquées par une pathologie de surface oculaire comme les dysfonctionnements glandulaires meibomiens, en particulier la rosacée oculaire, mais il s’agit d’un diagnostic d’élimination.
L’examen de la surface oculaire recherche enfin des anomalies cornéennes, comme une kératite sèche ou une kératite périphérique immunologique, ou des lésions conjonctivales à type d’érosion ou de nécrose.
Une accumulation de cholestérol accompagne le vieillissement de la sclère qui perd sa blancheur et jaunit. Les plaques sclérales hyalines sont parfois inflammatoires et peuvent mimer une scléromalacie perforante. Elles correspondent en fait à une métaplasie cartilagineuse au niveau des zones d’insertion des muscles droits latéraux. Elles sont visibles à l’œil nu sous la forme de zones ovalaires grisâtres d’environ 2 mm de grand axe vertical. Dans de rares cas, il se forme un séquestre qui peut tomber et laisser une dépression sclérale au fond de laquelle on devine la choroïde noire.
La classification des sclérites distingue les formes antérieures et postérieures, selon que l’inflammation sclérale siège respectivement en avant ou en arrière de l’ora serrata.
Les sclérites antérieures sont subdivisées en sclérites antérieures diffuses, nodulaires et nécrosantes avec ou sans inflammation (fig. 7-23), avec un pronostic de gravité croissante tant sur le plan local que général, les formes avec nécrose étant souvent associées à des vascularites systémiques potentiellement létales [5].
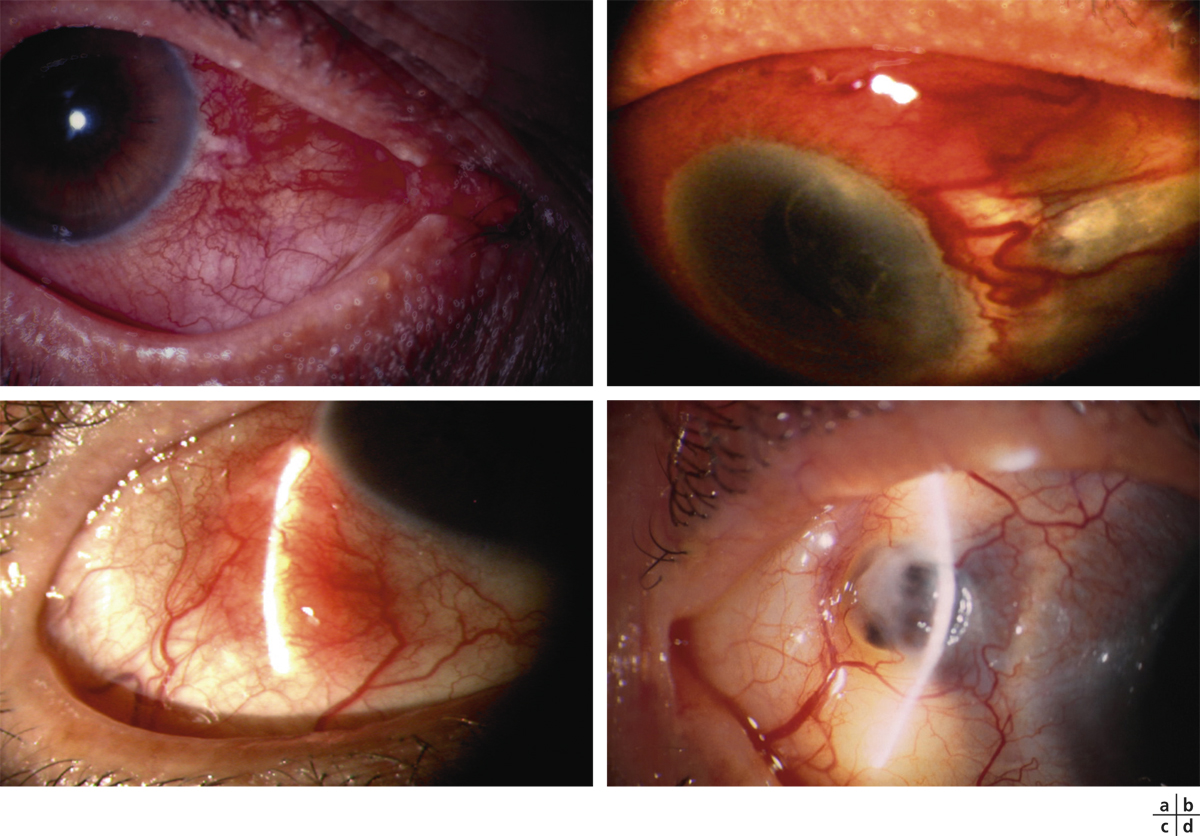
Fig. 7-23 Formes cliniques de sclérites antérieures : diffuse (a) ; nodulaire (b) ; nécrosante avec inflammation (c) ; nécrosante sans inflammation (d).
Les sclérites antérieures diffuses sont les plus fréquentes mais les moins sévères des inflammations sclérales. Elles débutent progressivement et de manière insidieuse. Elles récidivent volontiers et sont associées à des pathologies de système dans un peu moins de 50 % des cas [1]. Les pathologies généralement retrouvées, soit lors du bilan initial soit au cours du suivi, sont la polyarthrite rhumatoïde, les vascularites granulomateuses avec polyangéite (maladie de Wegener), les entérocolopathies, les arthrites réactionnelles et les spondylarthropathies (encadré 7-2).
Vascularites systémiques
• Polyarthrite rhumatoïde
• Péri-artérite noueuse
• Granulomatose de Wegener
• Syndrome de Churg-Strauss
• Cryoglobulinémie mixte
• Polychondrite atrophiante
• Maladie de Horton
• Vascularite de Cogan
• Syndrome de Behçet
Pathologies dermatologiques
• Lupus érythémateux disséminé
• Sarcoïdose
• Rosacée
Spondylarthropathies
• Spondylarthrite ankylosante
• Arthrite psoriasique
• Entérocolopathies (maladie de Crohn, rectocolite hémorragique)
Autres pathologies systémiques avec atteinte sclérale
• Vascularite à IgA (purpura rhumatoïde, maladie de Berger)
• Syndrome de Goodpasture
Granulomatose infectieuse
• Tuberculose
• Lèpre
• Syphilis
• Maladie de Lyme
Infections virales
• Herpes simplex virus
• Zona
• Hépatites B/C
• Cytomégalovirus
Iatrogénie
• Post-chirurgie (strabisme, chirurgie du ptérygion)
• Biphosphonates
• Erlotinib
• Topiramate
• Étanercept
• 5-fluorouracile/mytomicine C
La sclérite nodulaire est caractérisée par l’importance de l’œdème localisé qui accompagne l’inflammation sclérale. Elle se manifeste par un nodule rouge sombre parfois violacé, très souvent localisé près du limbe, fixe et particulièrement douloureux à la pression. Son évolution est plus chronique que celle des formes antérieures diffuses avec des récidives plus fréquentes et un risque de passage vers une sclérite nécrosante dans près de 10 % des cas. Le caractère nodulaire ne préjuge en rien de la physiopathologie granulomateuse ou non de la pathologie causale. Cependant cette sclérite a une sévérité locale plus importante que la forme diffuse et est plus volontiers associée qu’elle à des vascularites systémiques (dans près de 60 % des cas). Les récidives laissent fréquemment des cicatrices sclérales sombres qui sont plus souvent liées à un réarrangement de la matrice sclérale, sans amincissement ni perte de tissu, qu’à une fonte sclérale laissant deviner la choroïde au travers (scléromalacie). Une sclérite nodulaire unilatérale cortico-dépendante ou résistante au traitement sans association retrouvée à une pathologie générale doit faire rechercher une étiologie herpétique ou zostérienne, particulièrement chez les patients de plus de 50 ans [6]. Dans ces cas, l’association d’antiviraux et d’anti-inflammatoires est généralement efficace.
Les sclérites nécrosantes sont les plus rares mais les plus sévères tant sur le plan local que général. Ainsi, près d’un patient sur deux a une baisse significative de l’acuité visuelle et le taux de mortalité à 5 ans avoisine les 30 % [7]. La caractéristique clinique principale est la présence de zones blanches avasculaires entourées d’œdème scléral et de congestion des plexus vasculaires épiscléraux. La sclère atteinte s’amincit, devient transparente et laisse deviner la coloration sombre de la choroïde sous-jacente. La nécrose n’intéresse au début qu’une zone limitée. En l’absence de traitement adapté, celle-ci peut s’élargir et un séquestre scléral est à même de se former et se détacher de la surface oculaire. Une vascularite systémique est retrouvée dans près de 80 % des cas. La polyangéite granulomateuse de Wegener et la polyarthrite rhumatoïde sont les deux principales étiologies [8, 9]. Si une sclérite antérieure nécrosante avec inflammation peut révéler la première, il n’en est pas de même pour la seconde qui est généralement déjà connue et bien avancée quand l’atteinte oculaire survient. La sclérite nécrosante sans inflammation, aussi décrite sous le terme de scléromalacie perforante, ne s’accompagne ni de douleur ni de rougeur et est pathognomonique de la polyarthrite rhumatoïde évoluée [10, 11]. Pour certains, il s’agirait de l’extériorisation à travers la sclère de véritables nodules rhumatoïdes, mais cette assertion n’est pas confirmée par l’examen anatomo-pathologique qui retrouve seulement une inflammation intense avec infiltrats de cellules inflammatoires mononucléées et lésions de nécrose granulomateuse [1].
Des cas de sclérite nécrosante iatrogène sont rapportés après chirurgie oculaire [10]. On distingue les formes précoces, survenant dans les trois premières semaines postopératoires, des formes tardives diagnostiquées parfois de nombreuses années après une chirurgie de ptérygion ou de strabisme par exemple [11, 12]. Si des facteurs de retard de cicatrisation (diabète, iatrogénie locale, cryothérapie, antimitotiques, etc.) peuvent être retrouvés et incriminés dans la genèse de la sclérite, l’association à des pathologies systémiques est fréquente et doit être systématiquement recherchée. Des formes d’infections chroniques de la sclère ont aussi été décrites dans les suites parfois très lointaines de chirurgies oculaires [13, 14]. Dans ce contexte, cette complication doit être évoquée en cas de sclérite évolutive ne répondant pas aux traitements anti-inflammatoires usuels.
Une biopsie sclérale et/ou épisclérale avec examen anatomo-pathologique, microbiologique et immuno-histochimique à la recherche d’une vascularite est indiquée devant l’évolution défavorable sous traitement d’une sclérite nécrosante sans étiologie retrouvée. Elle permet de diagnostiquer des formes trompeuses de maladies comme un carcinome épidermoïde conjonctivo-épiscléral inflammatoire qui peut mimer une sclérite nécrosante [15, 16].
Les sclérites postérieures sont les inflammations sclérales situées en arrière de l’ora serrata. Leur diagnostic est particulièrement difficile, aussi bien lorsqu’elles sont isolées car l’œil n’est pas rouge, que lorsqu’elles sont associées à une sclérite antérieure qui pourrait faire oublier de la rechercher. Cela souligne l’importance de pratiquer un bilan ophtalmologique complet incluant l’examen du fond d’œil dans toute inflammation du segment antérieur oculaire. Une association à des pathologies systémiques est retrouvée dans 30 % des cas et est donc plus rare qu’avec les autres formes cliniques de sclérites [17]. La sévérité particulière des sclérites postérieures est avant tout liée aux atteintes oculaires contiguës : choroïde, procès ciliaires, rétine, nerf optique. Les signes cliniques évocateurs comprennent donc des plis choroïdiens ou rétiniens, un décollement de rétine exsudatif ou encore un œdème maculaire ou papillaire. L’angiographie fluorescéinique de la rétine recherchera : une vascularite avec diffusion du produit de contraste ; des décollements de l’épithélium pigmentaire et exsudatifs rétiniens ; une papillite. L’échographie en mode B oculaire peut aider au diagnostic en montrant en arrière le signe du T traduisant le refoulement des tissus orbitaires par l’œdème scléral et la capsule de Tenon autour du nerf optique [4]. L’imagerie par résonance magnétique (IRM) retrouve l’épaississement de la paroi oculaire postérieure et élimine une sinusite infectieuse à l’origine de la sclérite par contiguïté et pour laquelle une corticothérapie serait dangereuse. Le diagnostic différentiel est parfois difficile avec une tumeur choroïdienne, un décollement de l’épithélium pigmentaire et/ou de rétine inflammatoire dans le cadre d’une maladie de Vogt-Koyanagi-Harada, d’une effusion uvéale, etc. [4].
L’épisclérite est une inflammation localisée à l’épisclère et à la capsule de Tenon. L’épisclérite simple est diffuse, impliquant un ou plusieurs quadrants de la surface oculaire. L’épisclérite nodulaire est localisée, en relief. Les deux formes représentent le plus souvent une pathologie bénigne ne se compliquant pas sur le plan local. Rarement, elles sont associées à une pathologie systémique [18].
L’épisclérite simple est la plus fréquente et la plus bénigne des épisclérites. Contrairement à la sclérite dont le début est souvent progressif et douloureux, elle est caractérisée par la survenue brutale d’une rougeur vive et d’une gêne oculaire modérée. Elle s’oppose également à la sclérite par sa fugacité, sa guérison spontanée et la fréquence de ses récidives. L’épisclérite simple est le plus souvent idiopathique ou associée à des pathologies limitées à la surface oculaire. Sa guérison spontanée malgré des récidives et son caractère à bascule (tantôt un œil, tantôt l’autre) sont rassurants quant à l’absence de maladie générale sévère sous-jacente. Cependant si elle résiste au traitement conventionnel ou en présence d’autres symptômes ophtalmologiques ou systémiques, un bilan étiologique à la recherche de vascularite sera réalisé. Les spondylarthropathies s’accompagnent plus fréquemment d’épisclérite que de sclérite [19].
L’épisclérite nodulaire représente 30 % des épisclérites. Elle réalise une inflammation localisée de l’épisclère de début plus insidieux et de durée plus prolongée que la forme simple diffuse. Elle siège volontiers près de la cornée, plutôt dans l’axe horizontal et en nasal, et est souvent légèrement douloureuse. À la différence de la sclérite nodulaire, le nodule est mobilisable à l’aide du bord libre palpébral. Le test de vasoconstriction à l’épinéphrine est positif en quelques secondes. En outre, à l’examen biomicroscopique en fente fine, les lignes de profil de la conjonctive et de la sclère, encore plus visibles après le test précédent, apparaissent exagérément séparées. Les étiologies de l’épisclérite nodulaire ne diffèrent pas de celles de la forme simple diffuse si ce n’est la plus forte prévalence des pathologies de système [20].
En dehors des inflammations cornéennes par contiguïté aux sclérites périlimbiques, il en existe qui sont en relation avec la vascularisation limbique de type terminal propice aux dépôts de complexes immuns circulants. Ceux-ci sont à leur tour à l’origine d’une activation de toute la cascade inflammatoire immunologique de type III décrite par Gell et Coombs. Les vascularites systémiques peuvent ainsi se compliquer de kératites périphériques immunologiques indépendamment ou non d’une inflammation sclérale [21]. L’association d’une sclérite à cette pathologie cornéenne requiert un bilan clinique et paraclinique à la recherche d’une maladie systémique qui en est toujours la cause [22]. La maladie de Wegener et la polyarthrite rhumatoïde sont deux des principales étiologies à rechercher, avec les autres vascularites à anticorps anticytoplasme des polynucléaires neutrophiles (ANCA) et les entérocolopathies.
Différentes formes cliniques de complications cornéennes sont décrites au cours des sclérites. Les infiltrats cornéens périphériques, sans intervalle de cornée saine avec l’arcade vasculaire limbique, sont les plus typiques. Ils peuvent se transformer en kératite ulcérée périphérique qui progresse de manière circonférentielle et centripète. C’est le pseudo-ulcère de Mooren par opposition à l’ulcère de Mooren qui n’est jamais associé à une sclérite et est considéré comme primitif (fig. 7-24).
Les autres inflammations cornéennes associées aux sclérites sont plus rares : amincissement périphérique sans ulcération ni infiltrat, comparable à une gouttière (furrow des Anglo-Saxons) ; sclérokératite se caractérisant par un infiltrat périphérique inflammatoire et lipidique non ulcéré très dense associé à des néovaisseaux profonds. Cette dernière ressemble à une kératite interstitielle tellement blanche qu’elle donne l’impression que la sclère se prolonge dans la cornée [23, 24].
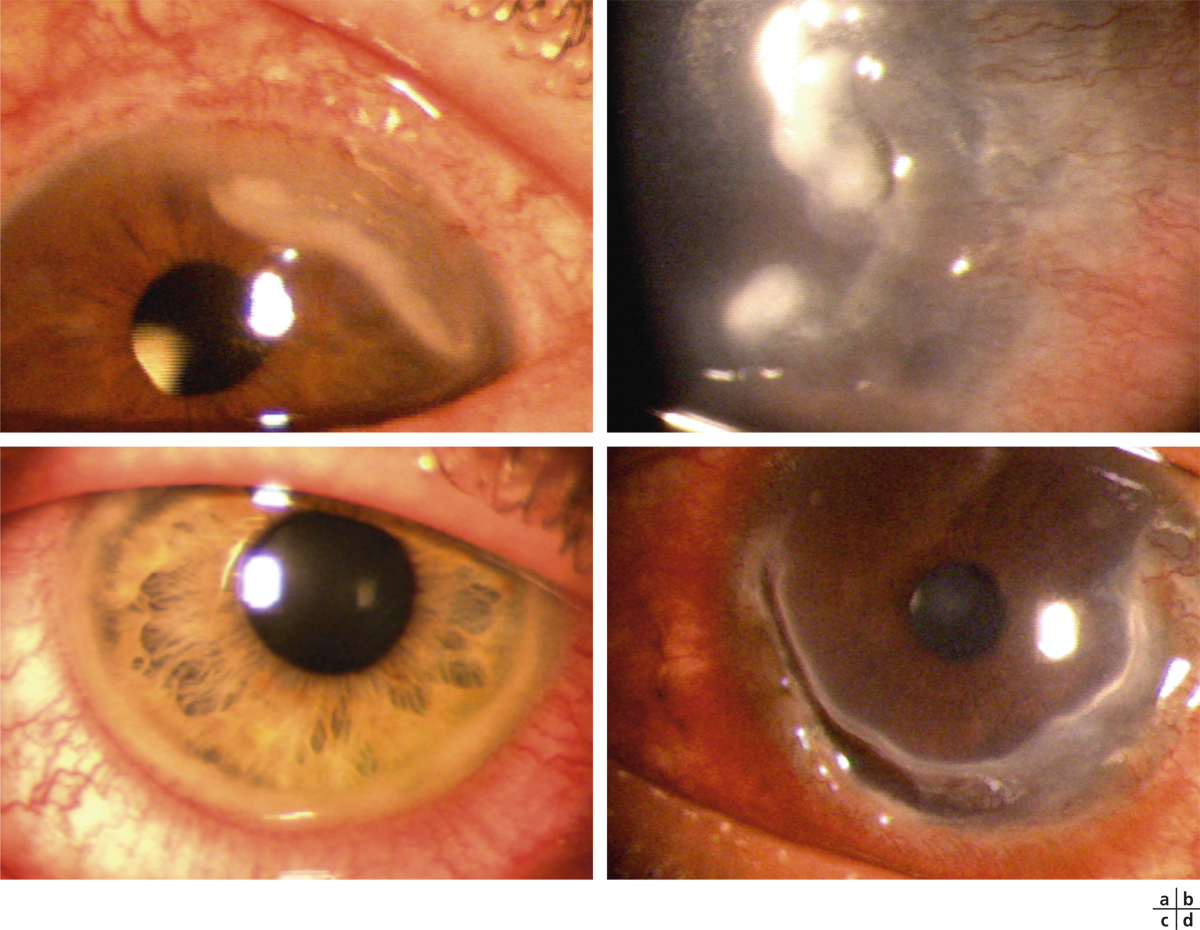
Fig. 7-24 Kératites périphériques immunologiques.
Évolution vers l’ulcération (a, b). Progression circonférentielle (c) et centripète (d).
La prise en charge diagnostique des patients atteints d’inflammation sclérale rassemble des investigations spécialisées d’ordre ophtalmologique, rhumatologique et de médecine interne. Avant d’installer le patient à la lampe à fente, une inspection générale recherchera aussi bien des difficultés à la marche que des déformations digitales ou chondrales au niveau du nez et des oreilles. L’interrogatoire portera avant tout sur les affections articulaires, cutanées et gastro-entérologiques.
L’examen ophtalmologique à l’œil nu puis à la lampe à fente doit être complet, à la recherche d’anomalies palpébrales (vésicules, blépharites, cicatrices) et de la surface oculaire en dehors de la sclérite, et d’une inflammation endoculaire. En l’absence de sclérite nécrosante, des ulcérations conjonctivales peuvent être dues à un herpèsvirus, une maladie de Behçet ou une péri-artérite noueuse [6, 25, 26].
Le bilan biologique à la recherche d’une pathologie systémique méconnue (encadré 7-3)
• Numération formule sanguine (NFS), vitesse de sédimentation (VS), C reactive proteine (CRP), fibrinogène, électrophorèse des protéines plasmatiques
• Bilan rénal et hépatique
• Uricémie
• Bandelette urinaire (protéinurie, hématurie)
• Réaction au latex–Waler-Rose
• Anticorps anticitrulline
• Anticorps anti-Ro-SSA, anti-La-SSB
• ± Biopsie des glandes salivaires accessoires
• Anticorps antinucléaires ± anti-ADN natifs
• Anticorps anticytoplasme des polynucléaires neutrophiles (ANCA)
• TPHA (treponema pallidum haemagglutination assay), VDRL (venereal disease research laboratory)
• Sérologie VIH (1 et 2)
• Sérologies hépatites B et C
• intradermoréaction (IDR) : tuberculine/quantiféron
• Imagerie
• Radiographie de thorax (face et profil)
• Radiographies des articulations douloureuses
sera réalisé dès la première poussée de sclérite et d’épisclérite nodulaire et lors d’épisclérites simples résistantes et/ou récidivantes. Lin et al. recommandent de rechercher une polyarthrite rhumatoïde et une maladie de Wegener, même chez des patients asymptomatiques, car ils ont retrouvé un facteur rhumatoïde et des ANCAs positifs dans respectivement 30 % et 10 % des cas, et environ la moitié de ces patients ont déclaré la maladie dans les 5 ans. En cas de bilan biologique négatif, le risque de développement de la maladie était inférieur à 5 %. Les résultats de cette étude soulignent l’intérêt d’une bonne surveillance et de savoir renouveler les bilans en cas de sclérite rebelle et sans étiologie [27]. Chez les patients ayant déjà une pathologie systémique connue au moment de la déclaration de la sclérite, une recherche d’évolutivité de la maladie doit être effectuée.
L’échographie en mode B est l’examen de prédilection pour le diagnostic des sclérites postérieures. Elle permet de quantifier l’épaississement scléral et de distinguer les sclérites postérieures diffuses des formes nodulaires. Elle est enfin essentielle pour le diagnostic différentiel avec les tumeurs du pôle postérieur. L’IRM et le scanner orbitaire peuvent être utiles, non seulement pour confirmer l’épaississement scléral postérieur, mais aussi pour apprécier l’orbite et les sinus. L’optical coherence tomography (OCT) et l’angiographie fluorescéinique du pôle postérieur recherchent des modifications de la perfusion sclérale et les conséquences de l’inflammation sur la choroïde, la rétine et la papille optique.
L’angiographie de segment antérieur au vert d’indocyanine permet d’objectiver la vascularite au niveau des plexus épiscléraux. Dans la sclérite, le colorant diffuse hors des vaisseaux et s’accumule au niveau de la sclère. Des zones d’ischémie des plexus épiscléraux profonds, parfois non identifiables à la lampe à fente, peuvent être révélées en angiographie sous la forme de modifications du calibre vasculaire ou d’aires de non-perfusion. La valeur pronostique de ces signes angiographiques reste encore à définir [28–31].
Les carcinomes de la conjonctive peuvent passer pour une sclérite nécrosante, comme les deux cas que nous avons rapportés et qui comportaient aussi une atteinte cornéenne périphérique. Des lymphomes oculaires de type MALT (mucosa-associated lymphoid tissue) empruntent également parfois un aspect de sclérite [32, 33].
Les angiomes épiscléraux et les autres anomalies de la vascularisation sclérale, comme celles secondaires à des fistules durales ou carotido-caverneuses, sont reconnus rapidement dans la majorité des cas [16].
L’inflammation d’une pinguecula et d’un nævus kystique bénin est fréquemment prise pour une épisclérite nodulaire. Dans ce dernier cas, la constatation de kystes intralésionnels aide à redresser le diagnostic.
Les plaques sclérales hyalines peuvent dans certains cas former un séquestre qui, après être expulsé, laisse parfois un défect douloureux. C’est un diagnostic différentiel de la scléromalacie perforante. Cependant, la situation typique à l’insertion d’un muscle droit horizontal, la présence de plaques sclérales hyalines typiques ipsilatérales et controlatérales ainsi que l’absence d’antécédents de polyarthrite rhumatoïde permettent de faire le diagnostic.
Certaines sclérites postérieures peuvent être confondues à l’examen du fond de l’œil avec une néoplasie (mélanome, métastase de la choroïde ou rétinoblastome envahissant la sclère), mais les examens d’imagerie redressent le diagnostic [34, 35].
Les anti-inflammatoires topiques font partie de l’arsenal thérapeutique des sclérites, mais sont rarement efficaces seuls. Si tel est le cas, cela traduit une inflammation sclérale bénigne ou une erreur diagnostique avec une épisclérite. Dans les formes sévères de sclérite nécessitant un traitement immunosuppresseur systémique, le traitement anti-inflammatoire local permettrait dans une certaine mesure une très relative épargne thérapeutique.
Les anti-inflammatoires non stéroïdiens (AINS) topiques sont fréquemment utilisés pour traiter les épisclérites en adjonction avec les traitements mouillants de la surface oculaire. En cas de syndrome de Gougerot-Sjögren, leur utilisation est déconseillée en raison du risque de complications cornéennes comme la kératolyse aseptique [11, 36, 37].
Les corticoïdes topiques sont le traitement de première intention de choix des sclérites antérieures peu sévères. Ils permettent la réalisation du bilan à la recherche d’une pathologie systémique sans interférer avec ses résultats. Dans les formes plus sévères, ils procurent une réponse anti-inflammatoire immédiate le temps de la mise en route des traitements systémiques dont l’efficacité est parfois retardée. Ils permettent enfin une certaine épargne thérapeutique en contribuant à limiter les doses cumulées de corticoïdes par voie générale [4, 38].
Les AINS systémiques sont très efficaces dans les sclérites antérieures diffuses et nodulaires. Ils soulagent en quelques jours les patients mais doivent néanmoins être prolongés 3 à 4 semaines. Une attention particulière doit être portée aux patients atteints de sclérites à cause d’une pathologie sous-jacente connue ou méconnue avec, par exemple, glomérulonéphrite ou entérocolopathie inflammatoire, favorisant alors une iatrogénie des AINS systémiques sous forme respectivement d’une insuffisance rénale ou de saignement ou perforation digestifs.
Les sclérites nodulaires sont particulièrement sensibles aux AINS systémiques. En l’absence de réponse favorable dans les jours qui suivent l’initiation du traitement, il est conseillé de passer aux anti-inflammatoires systémiques [39, 40].
Les corticoïdes représentent le traitement de deuxième intention en cas de résistance aux AINS ou de première intention des sclérites antérieures sévères et postérieures. Un traitement oral à la dose de 1 ou 1,5 mg/kg/jour est souvent suffisant pour contrôler l’inflammation des sclérites modérées et sévères. La décroissance de la posologie est classiquement progressive, sur 4 à 6 semaines dans les sclérites idiopathiques, et guidée par les atteintes systémiques dans les autres cas. En cas de cortico-résistance ou de dépendance à des doses quotidiennes supérieures à 10 mg, un traitement immunosuppresseur est très souvent nécessaire [4].
Les bolus intraveineux de corticoïdes sont indiqués comme traitement d’attaque des sclérites nécrosantes et des sclérites postérieures sévères ou antérieures associées à des complications cornéennes. Un relais par voie orale est généralement réalisé après trois à cinq séries quotidiennes de perfusions de 500 à 1 000 mg de méthylprednisolone.
Les injections sous-conjonctivales de corticoïdes locaux ont longtemps été déconseillées en raison du risque théorique, d’une part, de ne traiter que la part ophtalmologique d’une pathologie systémique potentiellement mortelle, d’autre part de transformer une sclérite simple ou nodulaire en sclérite nécrosante en raison de l’effet délétère des corticoïdes sur le renouvellement de la matrice extracellulaire [41, 42].
Des études récentes ont néanmoins démontré l’intérêt des injections sous-conjonctivales de faibles doses de triamcinolone pour réduire la cortico-dépendance systémique de certaines formes de sclérites non nécrosantes. Une surveillance attentive reste nécessaire à cause des risques infectieux et pour ne pas mésestimer l’évolutivité d’une pathologie systémique [43–46].
De nombreux traitements peuvent être utilisés pour limiter les doses ou potentialiser l’effet des corticoïdes chez les patients atteints de sclérites sévères. Certains immunosuppresseurs, dont le cyclophosphamide, sont particulièrement adaptés aux formes sévères nécrosantes et/ou associées à des kératites périphériques ulcérantes. D’autres molécules sont indiquées, plus particulièrement dans certaines étiologies, pour limiter la dépendance cortisonée. C’est le cas de la disulone pour la polychondrite atrophiante, du méthotrexate et des anti-TNF (anti-tumor necrosis factor) pour la polyarthrite rhumatoïde et du rituximab pour la vascularite de Wegener [47, 48–50].
La prise en charge thérapeutique des ulcères cornéens de type pseudo-Mooren est avant tout médicale. Dans les formes associées à des ulcérations évolutives, la résection conjonctivale en regard des lésions cornéennes a été proposée pour limiter l’infiltration inflammatoire à ce niveau. Les greffes de membrane amniotique ont progressivement remplacé l’application de colle cyanoacrylate qui était auparavant déposée sur le fond de l’ulcération. En cas de perforation cornéenne imminente ou avérée, une kératoplastie lamellaire cornéosclérale peut être réalisée dans un but architectonique et immunologique, le greffon avasculaire limitant la propagation de l’inflammation sclérale. De même au cours des sclérites nécrosantes, des patchs scléraux s’avèrent parfois utiles [16, 26, 51–54].
[1] Sainz de la Maza M, Tauber J, Foster CS. The Sclera. Boston : Springer US ; 2012.
[2] Sainz de la Maza M, Molina N, Gonzalez-Gonzalez LA, et al. Scleritis therapy. Ophthalmology 2012 ; 119 : 51-8.
[3] Gabison E, Hoang-Xuan T. Scleritis : when might a systemic disease be involved ? J Fr Ophtalmol 2010 ; 33 : 593-8.
[4] Gabison EE, Hoang-Xuan T. Sclérites, épisclérites et autres pathologies de la sclère. Encycl Méd Chir (Elsevier, Paris). Ophtalmologie, 21-210-A-10. 2008 : p. 1-10.
[5] Foster CS. Ocular manifestations of the potentially lethal rheumatologic and vasculitic disorders. J Fr Ophtalmol 2013 ; 36 : 526-32.
[6] Gonzalez-Gonzalez LA, Molina-Prat N, Doctor P, et al. Clinical features and presentation of infectious scleritis from herpes viruses : a report of 35 cases. Ophthalmology 2012 ; 119 : 1460-4.
[7] Foster CS, Forstot SL, Wilson LA. Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripheral ulcerative keratitis. Effects of systemic immunosuppression. Ophthalmology 1984 ; 91 : 1253-63.
[8] Sainz de la Maza M, Foster CS, Jabbur NS. Scleritis associated with systemic vasculitic diseases. Ophthalmology 1995 ; 102 : 687-92.
[9] Watkins AS, Kempen JH, Choi D, et al. Ocular disease in patients with ANCA-positive vasculitis. J Ocul Biol Dis Infor 2009 ; 3 : 12-9.
[10] Sainz de la Maza M, Jabbur NS, Foster CS. Severity of scleritis and episcleritis. Ophthalmology 1994 ; 101 : 389-96.
[11] Machado O, Curi A, Fernandes RS, et al. Scleritis : clinical characteristics, systemic associations, treatment and outcome in 100 patients. Arq Bras Oftalmol 2009 ; 72 : 231-5.
[12] Doshi RR, Harocopos GJ, Schwab IR, et al. The spectrum of postoperative scleral necrosis. Surv Ophthalmol 2013 ; 58 : 620-33.
[13] Aydin A, Aksoy Y, Unal MH, et al. Necrotizing scleritis after pterygium surgery using mitomycin C. J Fr Ophtalmol 2012 ; 35 : 74-5.
[14] Huang CY, Lin HC, Yang ML. Necrotizing scleritis after strabismus surgery in thyroid eye disease. JAAPOS 2013 ; 17 : 535-6.
[15] Hodson KL, Galor A, Karp CL, et al. Epidemiology and visual outcomes in patients with infectious scleritis. Cornea 2013 ; 32 : 466-72.
[16] Yamazoe K, Shimazaki-Den S, Otaka I, et al. Surgically induced necrotizing scleritis after primary pterygium surgery with conjunctival autograft. Clin Ophthalmol Auckl NZ 2011 ; 5 : 1609-11.
[17] Lin HC, Ma DHK, Chen YF, et al. Late-onset intrascleral dissemination of Stenotrophomonas maltophilia scleritis after pterygium excision. Cornea 2011 ; 30 : 712-5.
[18] Sainz de la Maza M, Foster CS, Jabbur NS. Scleritis associated with rheumatoid arthritis and with other systemic immune-mediated diseases. Ophthalmology 1994 ; 101 : 1281-6.
[19] Fong LP, Sainz de la Maza M, Rice BA et al. Immunopathology of scleritis. Ophthalmology 1991 ; 98 : 472-9.
[20] Watson P, Romano A. The impact of new methods of investigation and treatment on the understanding of the pathology of scleral inflammation. Eye Lond Engl 2014 ; 30 : 100.
[21] Wakefield D, Di Girolamo N, Thurau S, et al. Scleritis : immunopathogenesis and molecular basis for therapy. Prog Retin Eye Res 2013 ; 35 : 44-62.
[22] Cordero-Coma M, García-Morán A, Yilmaz T, et al. Adjunctive globe magnetic resonance imaging in the diagnosis of posterior scleritis. Can J Ophthalmol 2011 ; 46 : 329-32.
[23] Garza-Leon M, Flores D, Alarcón-Galván G, Sánchez-Martínez C. Bilateral scleritis and sclerokeratitis associated with IgA nephropathy. J Ophthalmic Inflamm Infect 2012 ; 2 : 207-10.
[24] Khan IJ, Barry RJ, Amissah-Arthur KN, et al. Ten-year experience of pulsed intravenous cyclophosphamide and methylprednisolone protocol (PICM protocol) in severe ocular inflammatory disease. Br J Ophthalmol 2013 ; 97 : 1118-22.
[25] Khairallah M, Accorinti M, Muccioli C, et al. Epidemiology of Behçet disease. Ocul Immunol Inflamm 2012 ; 20 : 324-35.
[26] Akova YA, Jabbur NS, Foster CS. Ocular presentation of polyarteritis nodosa. Clinical course and management with steroid and cytotoxic therapy. Ophthalmology 1993 ; 100 : 1775-81.
[27] Sainz de la Maza M, Molina N, Gonzalez-Gonzalez LA, Doctor PP, et al. Clinical characteristics of a large cohort of patients with scleritis and episcleritis. Ophthalmology 2012 ; 119 : 43-50.
[28] Lin P, Bhullar SS, Tessler HH, et al. Immunologic markers as potential predictors of systemic autoimmune disease in patients with idiopathic scleritis. Am J Ophthalmol 2008 ; 145 : 463-71.
[29] Lemarinel B, Gabison E, Doan S, et al. Anterior-segment indocyanine green angiography in the management of anterior scleritis. J Fr Ophtalmol 2008 ; 31 : 495-501.
[30] Guex-Crosier Y, Durig J. Anterior segment indocyanine green angiography in anterior scleritis and episcleritis. Ophthalmology 2003 ; 110 : 1756-63.
[31] Herbort CP, Bodaghi B, Lehoang P. Indocyanine green angiography in ocular inflammatory diseases : principles, schematic interpretation, semiology and clinical value. J Fr Ophtalmol 2001 ; 24 : 423-47.
[32] Hoang-Xuan T, Bodaghi B, Toublanc M, et al. Scleritis and mucosal-associated lymphoid tissue lymphoma : a new masquerade syndrome. Ophthalmology 1996 ; 103 : 631-5.
[33] Gaucher D, Bodaghi B, Charlotte F, et al. MALT-type B-cell lymphoma masquerading as scleritis or posterior uveitis. J Fr Ophtalmol 2005 ; 28 : 31-8.
[34] Kafkala C, Daoud YJ, Paredes I, et al. Masquerade scleritis. Ocul Immunol Inflamm 2005 ; 13 : 479-82.
[35] Yakoubi S, Touzani F, Knani L, et al. Senile scleral plaque associated with scleritis in a patient with rheumatoid arthritis. J Fr Ophtalmol 2012 ; 35 : 625-33.
[36] Sainz de la Maza M, Jabbur NS, Foster CS. An analysis of therapeutic decision for scleritis. Ophthalmology 1993 ; 100 : 1372-6.
[37] Sainz de la Maza M, Molina N, Gonzalez-Gonzalez LA, et al. Scleritis therapy. Ophthalmology 2012 ; 119 : 51-8.
[38] Rachitskaya A, Mandelcorn ED, Albini TA. An update on the cause and treatment of scleritis. Curr Opin Ophthalmol 2010 ; 21 : 463-7.
[39] Leal C, Le Roux K, Rahmi A, et al. Scleritis, clinical features, etiologies and treatment : a case series of 32 patients. Rev Med Interne 2014 ; 35 : 491-7.
[40] Wieringa WG, Wieringa JE, ten Dam-van Loon NH, et al. Visual outcome, treatment results, and prognostic factors in patients with scleritis. Ophthalmology 2013 ; 120 : 379-86.
[41] Charkoudian LD, Ying G, Pujari SS, et al. High-dose intravenous corticosteroids for ocular inflammatory diseases. Ocul Immunol Inflamm 2012 ; 20 : 91-9.
[42] Berchicci L, Miserocchi E, Di Nicola M, et al. Clinical features of patients with episcleritis and scleritis in an Italian tertiary care referral center. Eur J Ophthalmol 2014 ; 24 : 293-8.
[43] Albini TA, Zamir E, Read RW, et al. Evaluation of subconjunctival triamcinolone for nonnecrotizing anterior scleritis. Ophthalmology 2005 ; 112 : 1814-20.
[44] Sohn EH, Wang R, Read R, et al. Long-term, multicenter evaluation of subconjunctival injection of triamcinolone for non-necrotizing, noninfectious anterior scleritis. Ophthalmology 2011 ; 118 : 1932-7.
[45] Roufas A, Jalaludin B, Gaskin C, et al. Subconjunctival triamcinolone treatment for non-necrotising anterior scleritis. Br J Ophthalmol 2010 ; 94 : 743-7.
[46] Johnson KS, Chu DS. Evaluation of sub-Tenon triamcinolone acetonide injections in the treatment of scleritis. Am J Ophthalmol 2010 ; 149 : 77-81.
[47] Bernauer W, Pleisch B, Brunner M. Five-year outcome in immune-mediated scleritis. Graefes Arch Clin Exp Ophthalmol 2014 ; 10 : 23-8.
[48] Miserocchi E, Pontikaki I, Modorati G, et al. Anti-CD 20 monoclonal antibody (rituximab) treatment for inflammatory ocular diseases. Autoimmun Rev 2011 ; 11 : 35-9.
[49] Saadoun D, Bodaghi B, Bienvenu B, et al. Biotherapies in inflammatory ocular disorders: Interferons, immunoglobulins, monoclonal antibodies. Autoimmun Rev 2013 ; 12 : 774-83.
[50] Hoang-Xuan T, Foster CS, Rice BA. Scleritis in relapsing polychondritis. Response to therapy. Ophthalmology 1990 ; 97 : 892-8.
[51] Choi W, Lee SS, Park YG, et al. A case of necrotizing keratoscleritis in primary Sjogren’s syndrome. Korean J Ophthalmol 2011 ; 25 : 275-7.
[52] Van der Horst-Bruinsma IE, Lems WF, Dijkmans B. A systematic comparison of rheumatoid arthritis and ankylosing spondylitis. Clin Exp Rheumatol 2009 ; 27 : S43–49.
[53] Akpek EK, Uy HS, Christen W, et al. Severity of episcleritis and systemic disease association. Ophthalmology 1999 ; 106 : 729-31.
[54] Stylianides A, Jones MNA, Stewart RMK, et al. Rheumatoid Arthritis–associated corneal ulceration : mortality and graft survival. Ophthalmology 2013 ; 120 : 682-6.