

F. Maÿ
La constatation d’une pression intra-oculaire (PIO) élevée doit conduire à rechercher les signes d’une dispersion pigmentaire et à réaliser une gonioscopie.
La dispersion pigmentaire est plus fréquente chez les jeunes myopes de sexe masculin.
La trabéculopathie pigmentaire est à l’origine de l’hypertension intra-oculaire pigmentaire, puis de la conversion vers un glaucome pigmentaire.
Le glaucome pigmentaire survient dans environ 10 à 20 % des dispersions pigmentaires.
Le glaucome pigmentaire (GP) caractérise une neuropathie optique glaucomateuse secondaire à un syndrome de dispersion pigmentaire récent ou ancien.
Le syndrome de dispersion pigmentaire (DP) survient sur une configuration anatomique oculaire particulière et se caractérise par une libération de pigment, provenant de l’épithélium postérieur de l’iris, qui se disperse et se dépose sur tous les éléments du segment antérieur du globe oculaire.
L’hypertension intra-oculaire pigmentaire (HTP) est définie par une pression intra-oculaire supérieure à 21 mmHg, présente au cours ou au décours d’un syndrome de dispersion pigmentaire, sans retentissement anatomique ou fonctionnel sur le nerf optique.
Le GP est une forme évolutive survenant dans 15 à 25 % des cas de DP. Il est caractérisé par d’importantes variations de la PIO. Si les altérations structurales et fonctionnelles sont semblables à celles du glaucome primitif à angle ouvert (GPAO), il présente cependant des modalités évolutives particulières importantes à connaître. La stratégie thérapeutique tiendra compte du stade de gravité du GP et aussi de l’existence ou non d’une dispersion active associée.
Les premières observations concernant le syndrome de DP remontent à plus d’un siècle. Krukenberg décrivit en 1899 les dépôts de pigment rétrocornéens en forme de fuseaux verticaux [22]. En 1901, Von Hippel [52] proposa l’hypothèse selon laquelle l’élévation pressionnelle est la conséquence de l’obstruction du trabéculum par le pigment, et Levinsohn [28] suggéra en 1909 que le pigment est d’origine irienne. La première description complète du GP revient à Sugar et Barbour en 1949 [50] et, par la suite, Sugar [51] publia une série de 147 cas en 1966. Richardson et al. [40] démontrèrent et décrivirent en 1977 la phagocytose des pigments de mélanine par les cellules endothéliales trabéculaires.
La théorie mécanique énoncée par Campbell en 1979 [8] est actuellement bien admise : la DP résulte du frottement de l’épithélium pigmenté de l’iris contre les fibres zonulaires lorsque la base de l’iris est convexe en arrière. En 1992, Karickhoff définit le concept de blocage pupillaire inverse [20].
Typiquement , la dispersion pigmentaire concerne les jeunes myopes de sexe masculin. Les études épidémiologiques menées aux États-Unis ont montré que la prévalence s’élevait à 2,45 %, avec une incidence de 4,8/100 000 par an [42, 49]. La DP paraît moins fréquente chez le non-Caucasien, mais est aussi moins facilement mise en évidence dans le cas d’iris épais et fortement pigmenté, et donc probablement sous-estimée. Elle est légèrement plus fréquente chez l’homme (58 à 67 % des DP) et chez les myopes (38 à 100 % selon les études). Généralement bilatérale et souvent asymétrique, elle apparaît entre 20 et 30 ans, évolue pendant quelques années puis s’amenuise après 50 ans [32, 43, 49, 51].
La PIO est le plus souvent normale, même dans les cas de DP typique. En effet, l’HTP est présente dans seulement environ 20 % des cas de DP [7]. Cette prévalence est difficile à apprécier car, dans les DP minimes ou débutantes, l’HTP est inconstante, la PIO ne s’élevant qu’en cas de libération de pigment. L’HTP prend un caractère permanent dès lors que les capacités du trabéculum sont dépassées ou après l’installation d’une trabéculopathie toxique.
Dans le monde occidental, le GP représente seulement 1 à 1,5 % des cas de glaucome mais il est la cause la plus fréquente de glaucome secondaire chez l’adulte jeune, en dehors des formes post-traumatiques [55]. Son incidence aux États-Unis s’élève à 1,4/100 000 par an. La plupart des patients atteints de GP sont des myopes de 30 à 50 ans, plus fréquemment de sexe masculin (78-93 %) [15, 32, 43, 49, 51].
En dehors de quelques formes familiales, la DP survient de façon apparemment sporadique, mais 4 à 21 % des patients présentent un antécédent familial [43, 49]. L’hérédité de la DP et du GP n’est pas encore totalement élucidée. Le premier gène associé à la DP a été localisé sur le chromosome 7, en 7q35-q36 par Andersen, en 1997 : le GPDS1 (glaucoma-related pigment dispersion syndrome 1) [4]. Un deuxième gène a été identifié sur le chromosome 18, en 18q21 [3]. Cependant, d’autres gènes pourraient être impliqués. La survenue d’un syndrome de DP résulterait en fait de l’association de la mutation de plusieurs gènes prédisposants et la conversion en GP nécessiterait l’implication associée d’autres gènes [25].
La libération de pigment de mélanine dans le segment antérieur résulte du frottement de la région périphérique de l’épithélium postérieur de l’iris contre les fibres zonulaires antérieures. Caprioli et al. [11] avaient démontré en 1986 que la chambre antérieure était plus profonde chez les patients présentant un syndrome de DP par rapport à ceux atteints de GPAO. Ce phénomène de frottement nécessite en effet une configuration anatomique oculaire particulière associant une chambre antérieure profonde (cas du myope), un angle iridocornéen largement ouvert, une insertion plus postérieure de l’iris, des procès ciliaires proéminents et un iridodonésis de la partie basale d’un iris fin, flottant et hyperlaxe.
Dans cette configuration anatomique, l’iris se plaque contre la face antérieure du cristallin sur une plus large zone de contact favorisant la survenue d’un bloc pupillaire inverse dans lequel l’iris se comporte comme une valve anti-retour, pompant l’humeur aqueuse de la chambre postérieure vers la chambre antérieure [20, 29]. L’hyperpression relative générée en chambre antérieure induit un bombement vers l’arrière de la moyenne périphérie de l’iris : il s’agit du recurvatum irien, bien mis en évidence par la tomographie en cohérence optique (OCT) du segment antérieur et la biomicroscopie ultrasonore (UBM) [38, 39]. L’instillation de pilocarpine tend la base de l’iris, éloigne l’épithélium postérieur de l’iris des fibres zonulaires et lève le bloc pupillaire inverse [7]. Celui-ci apparaît également dans des circonstances non pathologiques, lors du clignement, de l’accommodation et des mouvements oculaires, dans certaines positions de la tête et au cours de l’exercice physique [1, 20, 29]. La responsabilité de l’accommodation dans le bombement de l’iris vers l’arrière a été bien démontrée autant en UBM qu’en OCT de segment antérieur [1, 30, 38, 39].
Le pigment libéré à partir de l’épithélium postérieur de l’iris se déverse dans l’humeur aqueuse et va se déposer à la surface de tous les éléments du segment antérieur : cristallin, zonule, face antérieure de l’iris, endothélium cornéen et angle iridocornéen.
Les grains de mélanine sont piégés dans le trabéculum où ils s’accumulent dans les portions uvéales et cornéosclérales [2, 35]. Le pigment est progressivement phagocyté par les cellules endothéliales trabéculaires puis évacué à travers le trabéculum juxta-canaliculaire dans le canal de Schlemm [8, 40]. La PIO reste comprise dans des valeurs normales dans la plupart des syndromes de DP. Dans certains cas, les capacités du trabéculum seront dépassées : soit du fait d’une dispersion massive, soit par altération des cellules trabéculaires.
Lors d’une libération massive de pigment, après dilatation médicamenteuse ou après un effort physique, il se produit une obstruction brutale du trabéculum qui augmente la résistance à l’écoulement de l’humeur aqueuse, induisant un pic d’hypertension intra-oculaire. Une fois le pigment évacué, la PIO se normalise. Le processus est réversible.
Au cours de la phase initiale de la DP, le pigment est régulièrement phagocyté puis évacué. Si la dispersion se prolonge dans le temps, les capacités de phagocytose diminuent et les grains de mélanine s’accumulent dans les cellules endothéliales trabéculaires, conduisant à leur nécrose par effet toxique. Les cellules nécrosées sont évacuées par les macrophages, laissant à nu les lamelles trabéculaires qui fusionnent jusqu’à refermer les espaces trabéculaires. La trabéculopathie est constituée. Irréversible, elle devient responsable d’une élévation chronique de la PIO [2, 17, 40].
Il associe plusieurs anomalies spécifiques facilement identifiables à l’examen biomicroscopique.
Les plages d’atrophie de l’épithélium pigmenté de l’iris sont cunéiformes, à disposition radiaire, non confluentes et situées dans la moyenne périphérie de l’iris, dans le tiers moyen externe [8, 49]. Leur examen se fait en transillumination, à travers le stroma irien, la pupille étant éclairée de face. Difficiles à repérer dans les formes débutantes, il faut les rechercher dans le secteur nasal inférieur. Plus évidentes dans les iris fins et clairs, elles sont souvent masquées dans les iris épais (fig. 14-1).
En 1958, Scheie et Fleischhauer [44] décrivirent les cinq éléments constituant le syndrome de dispersion pigmentaire : le fuseau de Krukenberg, les fins dépôts de pigment sur la face antérieure de l’iris, la pigmentation du trabéculum, les plages atrophiques iriennes et les dépôts de pigment sur les fibres zonulaires ainsi que sur la capsule postérieure du cristallin près de l’équateur.
Le Tyndall pigmentaire traduit la suspension en chambre antérieure du pigment libéré à l’occasion d’une dilatation ou d’un effort physique, prenant l’aspect de décharges ou d’averses pigmentaires (« pigment showers »). Le pigment suit les mouvements de convexion de l’humeur aqueuse et se dépose à la surface de tous les éléments du segment antérieur.
Le fuseau de Krukenberg correspond au dépôt de pigment saupoudrant la face postérieure endothéliale de la partie centrale de la cornée. Typiquement, ce dépôt apparaît sous la forme d’un saupoudrage marron-brun de granules de mélanine se répartissant sur une bande à disposition verticale de 1-2 mm de large et 3-6 mm de hauteur, en forme de fuseau. L’aspect est moins évocateur dans les formes minimes qui nécessitent un examen en fente fine oblique. Le dépôt est alors moins dense, plus réduit en étendue, et prend l’aspect d’un très fin saupoudrage de grains de cannelle. Le fuseau de Krukenberg n’est pas toujours présent dans la DP ni strictement pathognomonique, mais il témoigne néanmoins d’une DP active (fig. 14-2 et 14-3 ; eFig 14-1).
Les dépôts sur la face antérieure de l’iris se présentent sous la forme d’un saupoudrage irrégulier « poivre et sel », constitué de pigment et de macrophages chargés de grains de mélanine, s’accumulant préférentiellement dans les cryptes et le long des plis concentriques périphériques. Si la dispersion est bilatérale et asymétrique, elle prend l’apparence d’une hétérochromie irienne.
La région moyenne périphérique de l’iris est généralement bombée en arrière. L’iris paraît fin, flasque, un peu trop grand, avec des plis concentriques et présentant fréquemment un iridodonésis au moindre clignement. La chambre antérieure est généralement profonde et l’angle iridocornéen largement ouvert (fig. 14-4). La profondeur de la chambre antérieure et le bombement de la base de l’iris vers l’arrière sont bien mis en évidence en OCT de segment antérieur ainsi qu’en UBM [24, 33]. Dans une étude portant sur 44 yeux présentant une DP ou un GP, Mora et al. [33] démontrent que les paramètres diagnostiques les plus performants en UBM sont la largeur de l’angle iridocornéen et la profondeur de la concavité irienne, mesurées en situation d’accommodation, en vision de près.
La pigmentation de l’angle iridocornéen concerne d’abord le trabéculum qui est anormalement bien visible et chargé par les grains de mélanine en partie phagocytés par les cellules endothéliales trabéculaires. Puis toutes les structures de l’angle sont progressivement recouvertes de pigment depuis l’anneau de Schwalbe jusqu’à la périphérie de l’iris. Dans certains cas, l’anneau de Schwalbe, souligné par la pigmentation, présente un aspect comparable à celui de la ligne de Sampaolesi dans le syndrome pseudo-exfoliatif. La pigmentation de l’angle iridocornéen lui confère une coloration brune prononcée et homogène, généralement plus marquée dans le sinus inférieur. Scheie propose une graduation de la pigmentation de l’angle iridocornéen avec une cotation de 0 à IV (fig. 14-5) [45].
Dans la chambre postérieure, le pigment se dépose sur la pars plana, la hyaloïde antérieure, les fibres zonulaires et l’équateur du cristallin [43, 44]. Les dépôts de pigment sur la capsule postérieure du cristallin ont été décrits pour la première fois par Zentmayer en 1938 [56]. L’accumulation de pigment sur la capsule postérieure le long des insertions des fibres zonulaires postérieures constitue les rayures de Scheie (fig. 14-6). La ligne de Scheie, quant à elle, traduit l’accumulation de pigment sur la capsule postérieure le long d’une ligne circulaire contournant le ligament de Wieger, zone annulaire de forte adhérence de la hyaloïde antérieure sur la capsule postérieure du cristallin [43, 44]. À la faveur d’une désinsertion du ligament de Wieger, le pigment peut s’infiltrer dans l’espace de Berger et se déposer sur la région centrale de la capsule postérieure, en s’accumulant de façon homogène et continue en nappe ou en plaque [36].
Des anomalies rétiniennes sont souvent rencontrées, telles que la dégénérescence grillagée, présente dans 20 à 33 % des cas. Comparativement aux myopes, les déchirures rétiniennes sont également plus fréquentes, 12 %, de même que les décollements de rétine, 5,5-6,6 % [43, 47, 53].

Fig. 14-1 Plages atrophiques de l’iris sur 360°.

Fig. 14-2 Fuseaux de Krukenberg.
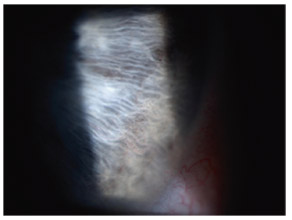
Fig. 14-3 Dépôts de pigment de mélanine sur la face antérieure de l’iris.
eFig. 14-1 Fuseaux de Krukenberg.

Fig. 14-4 Concavité de la base de l’iris en gonioscopie.

Fig. 14-5 Classification de la pigmentation de l’angle iridocornéen selon Scheie.

Fig. 14-6 Pigmentations zonulaires postérieures.
Dans les DP débutantes, le niveau de la PIO est très variable, alternant des pics consécutifs à des décharges pigmentaires et des phases de PIO normale. L’évolution de la PIO dépend de la fréquence des épisodes de libération de pigment, de leur importance et de l’état du trabéculum.
L’HTP apparaît de façon permanente lorsque le pigment, recouvrant complètement l’angle iridocornéen et obstruant durablement le trabéculum, s’oppose à l’excrétion de l’humeur aqueuse. Elle survient également comme conséquence de la trabéculopathie secondaire à l’effet toxique de l’excès de mélanine sur les cellules trabéculaires, le trabéculum devenant non fonctionnel. L’hypertonie pigmentaire apparaît, en moyenne, vers l’âge de 30 ans chez l’homme et un peu plus tard chez la femme, avec un décalage d’une dizaine d’années.
Typiquement, l’HTP se caractérise par des valeurs élevées de PIO. Siddiqui et al. [49] ont rapporté une valeur moyenne de 29 mmHg au moment du diagnostic. Dans l’étude de Migliazzo et al. [32], 25 % des HTP étaient supérieures à 31 mmHg et 12,5 % à 39 mmHg. La deuxième caractéristique de l’HTP est d’être variable au cours de la journée et d’un jour à l’autre, avec d’importantes variations, pouvant aller de 25 à 56 mmHg [51]. Ces pics pressionnels engendrent des signes fonctionnels fréquemment signalés par les patients : brouillard visuel, halos, céphalées, en particulier après un effort physique.
La DP est considérée comme active tant qu’il se produit une libération de pigment qui se manifeste par un Tyndall pigmentaire, un fuseau de Krukenberg, des dépôts sur la surface de l’iris, une pigmentation de l’angle et des pics pressionnels.
Avec le temps, la dispersion pigmentaire diminue puis s’épuise, souvent après 50 ans, soit par érosion de tout l’épithélium postérieur de l’iris en contact avec les fibres zonulaires, soit par l’augmentation de volume du cristallin avec l’âge qui éloigne l’épithélium postérieur de l’iris de la zonule, soit par le myosis sénile qui tend l’iris. La pigmentation de l’angle iridocornéen s’atténue très progressivement, et le sinus supérieur est souvent le dernier à s’éclaircir (pigment reversal sign). La réduction de l’encombrement trabéculaire diminue la résistance à l’évacuation de l’humeur aqueuse et conduit à une normalisation de la PIO [8, 32].
Il apparaît chez les sujets présentant une DP associée à une HTP prolongée responsable d’une neuropathie optique glaucomateuse. À ce titre, il apparaît bien comme un glaucome secondaire.
Les circonstances du diagnostic sont variables et peuvent se produire à différents stades de la maladie. La consultation est alors motivée par des symptômes fonctionnels et/ou l’observateur découvre à l’examen :
une DP active ;
une HTP associée à une DP active ;
une HTP sans DP active mais avec une pigmentation du trabéculum ;
des altérations glaucomateuses anatomiques ou fonctionnelles associées à des éléments orientant vers une DP ancienne ou encore active.
Typiquement, le GP associe :
des signes de la DP : plages atrophiques transilluminables de l’épithélium pigmenté irien, dépôts de pigment sur les éléments du segment antérieur, pigmentation de l’angle, présence d’un Tyndall pigmentaire et d’un fuseau de Krukenberg témoignant d’une DP active ;
une HTP avec des niveaux élevés et variables de la PIO ;
des altérations structurales du nerf optique : perte des fibres nerveuses rétiniennes, excavation papillaire, amincissement de l’anneau neurorétinien ;
des déficits périmétriques de nature glaucomateuse.
Les altérations structurales et fonctionnelles périmétriques sont parfaitement superposables à celles rencontrées dans le GPAO [19]. La grande similitude des atteintes glaucomateuses a longtemps conduit à classer le GP comme une forme clinique particulière du GPAO. Il s’en distingue cependant par sa rapidité d’évolution et son pronostic qui est généralement considéré comme sévère [32, 43]. L’évolutivité et la sévérité semblent en relation avec l’importance de la DP et avec le degré de pigmentation du trabéculum [15, 43].
Parallèlement à la diminution de la DP dans le temps, la PIO peut parfois se normaliser et laisser un tableau de GPAO [51]. C’est le GP « éteint » ou « burn-out glaucoma » (voir chapitre 14-III), qui peut être confondu avec un glaucome à pression normale s’il est découvert à ce stade (voir plus loin « Diagnostic différentiel »).
Parmi les sujets présentant une DP, 5 à 10 % développeront un GP cinq à six ans après le diagnostic, 15 % quinze ans après et 35 % trente-cinq ans après [32, 49]. Toutefois, les proportions sont très variables d’une étude à l’autre, même si elles sont randomisées. Certains facteurs de risque ont été identifiés et devront être recherchés afin d’adapter l’attitude thérapeutique.
L’existence d’antécédents familiaux de glaucome est notée dans 26 à 48 % des GP, mais il ne s’agit pas forcément d’antécédent de GP. La présence d’un antécédent familial de glaucome augmente davantage le risque de conversion plutôt que le développement d’une DP [49].
Si la DP concerne autant les deux sexes, le GP, lui, concerne plutôt le sujet masculin. Chez la femme, il apparaît dix ans plus tard et est volontiers plus sévère et agressif. Paradoxalement, le risque de conversion glaucomateuse d’une HTP est identique quel que soit le sexe [32].
Les autres facteurs de conversion sont le degré de myopie, l’existence d’un fuseau de Krukenberg et une PIO supérieure à 21 mmHg lors du diagnostic initial, risque le plus important [23, 31, 49].
D’autres affections oculaires sont responsables de DP ; elles sont souvent unilatérales et leur présentation est différente : absence de concavité irienne et de bloc pupillaire inverse. Cependant, les tableaux peuvent être trompeurs, surtout s’il existe une hypertonie oculaire [37].
Le syndrome exfoliatif (voir chapitre 14-II) est unilatéral dans la moitié des cas et concerne plutôt des sujets plus âgés, autour de la soixantaine. Si l’hypertonie est présente, la pigmentation du trabéculum est moins prononcée, plus hétérogène, et les plages d’atrophie de l’épithélium postérieur de l’iris sont plus proches de la pupille.
Une uvéite antérieure peut s’accompagner d’une libération de pigment secondaire à une altération inflammatoire de l’épithélium postérieur de l’iris. L’uvéite herpétique est une cause classique de plages de dépigmentation irienne associée à une élévation de la PIO, mais la localisation des zones atrophiques est différente.
Les altérations iriennes post-traumatiques peuvent causer une dispersion pigmentaire, une pigmentation trabéculaire et une élévation de la PIO. Cependant, la pigmentation est plus fine, plus noire, hétérogène en mottes et prédomine dans l’angle inférieur [39].
La libération de pigment peut survenir également après une lésion iatrogène de l’iris lors de la chirurgie du segment antérieur, ou même après une iridotomie.
Des dispersions pigmentaires ont été décrites chez le pseudo-phake, par frottement de l’iris contre un implant de chambre postérieure dont l’optique est trop antérieure, ainsi que dans le cas d’implantation en piggyback [12, 14]. Les zones d’atrophie de l’épithélium postérieur de l’iris sont alors localisées plutôt dans la région péripupillaire de l’iris et non en périphérie [14].
L’iris sénile associe également des zones atrophiques transilluminables et une pigmentation modérée du trabéculum, mais la présentation est différente.
Une dispersion pigmentaire peut également se rencontrer dans d’autres situations cliniques :
les tumeurs mélaniques : mélanome, mélanocytome, mélanose oculaire ;
Tyndall pigmentaire au cours du décollement de rétine rhegmatogène.
Enfin, quelques cas de fibres zonulaires antérieures allongées associées à une DP ont été décrits, mais sans concavité irienne. Ces longues fibres antérieures viennent s’insérer très en avant jusque dans la région centrale de la capsule antérieure. Le frottement de l’épithélium postérieur de l’iris libère du pigment qui se dépose entre les fibres zonulaires donnant un aspect de stries pigmentées. L’absence de concavité irienne est confirmée en biomicroscopie ultrasonore [34].
Le glaucome à pression normale est le principal diagnostic différentiel du GP éteint ou burn-out glaucoma. Les dépôts de pigment ont quasiment disparu, la pigmentation de l’angle iridocornéen s’est éclaircie ne laissant qu’une fine pigmentation résiduelle du trabéculum du sinus supérieur. Cependant, des éléments demeurent : la largeur de l’angle, la concavité irienne, les plages iriennes transilluminables et aussi la présence de rayures de Scheie, signe quasi pathognomonique d’une DP ancienne.
Le traitement a pour but de ralentir voire d’arrêter le processus de neuropathie optique glaucomateuse, en diminuant la PIO et en la maintenant à une valeur cible. La réduction de la PIO sera obtenue soit en diminuant la sécrétion d’humeur aqueuse par des hypotonisants locaux ou généraux, soit en améliorant son évacuation. Cette dernière nécessitera, outre des hypotonisants facilitant l’excrétion de l’HA tels que les analogues des prostaglandines, l’arrêt de la dispersion pigmentaire active par l’iridotomie et la facilitation de l’excrétion trabéculaire au moyen des trabéculoplasties et des interventions filtrantes.
Les myotiques, la pilocarpine en particulier, sont théoriquement les plus appropriés. La pilocarpine tend la périphérie de l’iris, s’oppose à son bombement en arrière et évite son contact avec la zonule. Son instillation empêche la dispersion pigmentaire et les symptômes fonctionnels d’une HTP brutale après un exercice physique [8, 38, 39]. Cependant, son usage au long cours entraîne des spasmes accommodatifs, avec des effets secondaires para-sympathico-mimétiques généraux comme la sécheresse buccale, et expose à un risque accru de décollement de rétine. La périphérie rétinienne doit être systématiquement vérifiée avant la prescription de pilocarpine. Elle n’est plus utilisée dans cette indication en raison de toutes ces contraintes et effets indésirables.
Les bêtabloquants, les α2-agonistes, les inhibiteurs de l’anhydrase carbonique et les analogues des prostaglandines peuvent être prescrits pour leur action hypotonisante mais n’exercent aucun effet spécifique sur la DP.
Les analogues des prostaglandines n’accentuent pas la DP et ne sont pas contre-indiqués, même en première intention.
Proposée pour la première fois par Kurwa en 1984, son efficacité a été confirmée par Campbell en 1991 [9, 10]. L’iridotomie périphérique a pour principe de supprimer le gradient de pression entre les chambres antérieure et postérieure, et d’éviter la constitution d’un bloc pupillaire inverse. L’équilibration pressionnelle résultant de l’iridotomie périphérique se traduit immédiatement par l’aplatissement de l’iris qui ne bombe plus en arrière, évitant alors tout contact avec la zonule et stoppant ainsi le processus de dispersion pigmentaire [13, 20, 21, 38]. Cet aplatissement de l’iris après iridotomie périphérique est bien visible en UBM et en OCT de segment antérieur [13, 24].
En pratique, l’iridotomie périphérique est facile à réaliser car l’iris est mince et le laser Nd-YAG suffit habituellement (voir chapitre 16-I paragraphe « Traitements physiques ») (vidéo 14-1). La perforation se signale immédiatement par un aplanissement visible de l’iris. L’iridotomie périphérique est élargie de façon à obtenir un trou irien de pleine épaisseur, de taille suffisante, supérieur à 100 μm, mais pas trop large, inférieur à 500 μm, pour éviter un relargage trop massif de pigment. L’iridotomie périphérique est suivie d’un traitement local associant un anti-inflammatoire et surtout un hypotonisant, α2-agoniste, pendant quelques jours afin de limiter le pic pressionnel qui peut se révéler élevé et prolongé.
L’iridotomie périphérique est indiquée dès lors qu’il existe une DP active. En effet, elle interrompt le processus de libération de pigment. En revanche, elle n’exerce aucune action directe sur la PIO dont l’évolution dépend de l’existence ou non d’une trabéculopathie. Si les altérations trabéculaires sont réversibles, l’HTP peut s’amender dans le temps et la PIO se normaliser progressivement [16]. Inversement, l’iridotomie périphérique ne peut rien contre la trabéculopathie irréversible responsable de l’hypertonie chronique et ne permet pas d’éviter une conversion vers un GP [46].
La trabéculoplastie au laser à l’argon (TLA), proposée par Wise en 1979, est reconnue pour son efficacité à réduire l’HTP de façon significative et durable [41, 54]. Exerçant un effet photothermique, elle induit une rétraction tissulaire élargissant les espaces trabéculaires. L’HTP représente la meilleure indication de la TLA, avec une efficacité obtenue avec des impacts moins nombreux et d’intensité plus faible que dans le GPAO (voir chapitre 16-I paragraphe « Traitements physiques »). Son action est plus marquée chez le sujet jeune, effet peut-être lié à la plus grande capacité des cellules du sujet jeune à répondre à la stimulation [41, 48].
Proposée par Latina et Park en 1998, elle utilise un Laser Nd-YAG pulsé de 532 nm, délivrant des impacts de 400 nm de diamètre et de durée très brève, de l’ordre de 3 nanosecondes [6, 26, 27] (voir chapitre 16-I paragraphe « Traitements physiques »). En cas de forte pigmentation, il est recommandé de commencer par de faibles puissances et de procéder en plusieurs fois, par petites étendues, afin d’éviter les pics pressionnels [18]. L’HTP est une des meilleures indications de la trabéculoplastie sélective. Son efficacité est comparable à celle de la TLA, et elle s’estompe également progressivement avec le temps comme le rapportent Ayala et al. [5], qui montrent 85 % de succès à un an, 67 % à deux ans et 44 % à trois ans. En revanche, moins agressive pour le trabéculum, elle présente l’avantage d’autoriser des retraitements permettant de contrôler la PIO de façon prolongée.
L’iridectomie chirurgicale n’a pas d’indication dans le DP et le GP.
Les chirurgies filtrantes, trabéculectomie et sclérectomie profonde non perforante, restent un recours efficace, pour lever la résistance à l’écoulement de l’humeur aqueuse causée par la trabéculopathie pigmentaire (voir chapitre 16-I paragraphe « Traitements chirurgicaux »). Lors de la trabéculectomie externe, la pigmentation rend le trabéculum cribriforme plus visible et facilite son pelage. Le glaucome pigmentaire reste une des meilleures indications de la sclérectomie profonde non perforante, avec une efficacité durable.
La démarche thérapeutique se déduit de la physiopathogénie de la dispersion pigmentaire et s’aligne sur celle de la prise en charge du GPAO, avec quelques particularités (tableau 14-1).
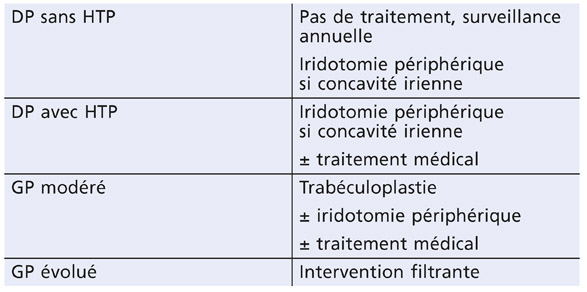
Tableau 14.1 – Recommandations thérapeutiques en fonction du stade évolutif.
En cas de dispersion active avec HTP ou avec des pics pressionnels élevés, l’iridotomie périphérique est indiquée et suivie d’un traitement hypotonisant. Si la PIO est normale, l’iridotomie est facultative à condition qu’une surveillance régulière soit possible.
Lorsque la DP est devenue inactive, l’iridotomie périphérique n’est plus nécessaire. Le traitement hypotonisant est prescrit en cas d’HTP. Sinon, une simple surveillance est suffisante.
Entre ces deux situations, il n’est pas facile de déterminer si la DP est active ou non, quand l’angle est modérément pigmenté, le bombement irien peu marqué, les zones d’atrophie mal visibles ou lorsque les dépôts rétrocornéens sont présents mais discrets. Dans ces situations, l’UBM peut se révéler utile pour mettre en évidence la position postérieure de l’iris, son bombement et son contact avec les fibres zonulaires.
L’HTP est caractérisée par des valeurs élevées et variables de PIO. Le traitement médical hypotonisant ne suffit pas à lui seul à normaliser la PIO, et il est généralement nécessaire de recourir à une iridotomie périphérique en cas dispersion active avec un iris concave associé ou à une trabéculoplastie sélective.
Le glaucome pigmentaire débutant requiert la normalisation de la PIO par hypotonisants médicaux ou par trabéculoplastie. L’iridotomie périphérique est indiquée si la DP est active. En cas d’échec du traitement médical ou laser, une intervention filtrante sera proposée.
En cas de GP sévère ou rapidement évolutif, l’intervention filtrante s’impose pour réduire significativement la PIO et éviter une aggravation des altérations du champ visuel, d’autant plus qu’il s’agit de sujets jeunes, myopes (facteur de risque), et que souvent le glaucome est découvert à un stade évolué.
Les glaucomes « éteints » ne nécessitent qu’une simple surveillance.
Retenir
Ne pas réaliser d’iridotomie périphérique systématique devant toute dispersion pigmentaire ou devant tout glaucome pigmentaire.
L’iridotomie périphérique est idéalement indiquée en cas de dispersion pigmentaire active avec hypertonie pigmentaire et iris concave.
Un glaucome étiqueté « glaucome à pression normale » peut en fait être un glaucome pigmentaire « éteint ».
Le glaucome pigmentaire est une bonne indication de trabéculoplastie et de chirurgie filtrante.
[1] Adam RS, Pavlin CJ, Ulanski LJ. Ultrasound biomicroscopic analysis of iris profile changes with accommodation in pigmentary glaucoma and relationship to age. Am J Ophthalmol. 2004 ; 138 : 652-54.
[2] Alvarado JA, Murphy CG. Outflow obstruction in pigmentary and primary open angle glaucoma. Arch Ophthalmol. 1992 ; 110 : 1769-78.
[3] Andersen JS, Parrish R, Greenfield D, et al. A second locus for the pigment dispersion syndrome and pigmentary glaucoma maps to 18q11-q21. Am J Hum Genet. 1998 ; 63 : A279.
[4] Andersen JS, Pralea AM, DelBono EA, et al. A gene responsible for the pigment dispersion syndrome maps to chromosome 7q35-q36. Arch Ophthalmol. 1997 ; 115 : 384-8.
[5] Ayala M. Long term outcomes of selective laser trabeculoplasty (SLT) treatment in pigmentary glaucoma. J Glaucoma. 2013 [Epub ahead of print].
[6] Barkana Y, Belkin M. Selective laser trabeculoplasty. Surv Ophthalmol. 2007 ; 52 : 634-54.
[7] Béchetoille A. Les glaucomes. Angers, éditions Japperenard, 2000.
[8] Campbell DG. Pigmentary dispersion and glaucoma : a new theory. Arch Ophthalmol. 1979 ; 97 : 1667-72.
[9] Campbell DG. Pigmentary glaucoma : mechanism and role for laser iridotomy. J Glaucoma. 1994 ; 3 : 173.
[10] Campbell DG. Pigmentary glaucoma. Humphrey Lecture. American Glaucoma Society Meeting, San Diego, CA, 1991.
[11] Caprioli J, Spaeth GL, Wilson RP. Anterior chamber depth in open angle glaucoma. Br J Ophthalmol. 1986 ; 70 : 831-6.
[12] Chang SHI, Lim G. Secondary pigmentary glaucoma associated with piggyback intraocular lens implantation. J Cataract Refract Surg. 2004 ; 30 : 2219-22.
[13] Chen MJ, Lin SC, Chen MJ. Effect of YAG laser iridotomy on intraocular pressure in pigmentary glaucoma. Br J Ophthalmol. 2002 ; 86 : 1443-4.
[14] Detry-Morel ML, Van Acker E, Pourjavan S, et al. Anterior segment imaging using optical coherence tomography and ultrasound biomicroscopy in secondary pigmentary glaucoma associated with in-the-bag intraocular lens. J Cataract Refract Surg. 2006 ; 32 : 1866-9.
[15] Farrar SM, Shields MB. Current concepts in pigmentary glaucoma. Surv Ophthalmol. 1993 ; 37 : 233-52.
[16] Gandolfi SA, Vecchi M. Effect of a YAG laser iridotomy on intraocular pressure in pigment dispersion syndrome. Ophthalmology. 1996 ; 103 : 1693-5.
[17] Gottanka J, Johnson DH, Grehn F, Lütjen-Drecoll E. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. J Glaucoma. 2006 ; 15 : 142-51.
[18] Harasymovycz PJ, Papamatheakis DG, Latina M, et al. Selective laser trabeculoplasty (SLT) complicated by intraocular pressure elevation in eyes with heavily pigmented trabecular meshworks. Am J Ophthalmol. 2005 ; 139 : 1110-3.
[19] Jonas JB, Dichtl A, Budde WM, Lang P. Optic disc morphology in pigmentary glaucoma. Br J Ophthalmol. 1998 ; 82 : 875-9.
[20] Karickhoff JR. Pigmentary dispersion syndrome and pigmentary glaucoma : a new mechanism concept, a new treatment, and a new technique. Ophthalmic Surg. 1992 ; 23 : 269-77.
[21] Karickhoff JR. Reverse pupillary block in pigmentary glaucoma : follow-up and new developments. Ophthalmic Surg. 1993 ; 24 : 562-3.
[22] Krukenberg F. Beiderseitige angeborene melanose der hornhaut. K Monatsbl Augenheilkd. 1899 ; 37 : 254-8.
[23] Küchle M, Mardin CY, Nguyen NX, et al. Quantification of aqueous melanin granules in primary pigment dispersion syndrome. Am J Ophthalmol. 1998 ; 126 : 425-31.
[24] Laemmer R, Mardin CY, Juenemann AG. Visualization of changes of the iris configuration after peripheral laser iridotomy in primary melanin dispersion syndrome using optical coherence tomography. J Glaucoma. 2008 ; 17 : 569-70.
[25] Lascaratos G, Shah A, Garway-Heath DF. The genetics of pigment dispersion syndrome and pigmentary glaucoma. Surv Ophthalmol. 2013 ; 58 : 164-75.
[26] Latina MA, Park C. Selective targeting of trabecular meshwork cells : in vitro studies of pulsed and CW laser interactions. Exp Eye Res. 1995 ; 60 : 359-72.
[27] Latina MA, Shibayan SA, Sliin DH, et al. Q-switched 532-nm Nd:YAG laser trabeculoplasty (selective laser trabeculoplasty). Ophthalmology. 1998 ; 105 : 1082-90.
[28] Levinsohn G. Beitrag zur pathologische anatomie aund pathologie des glaukoms. Arch Augenheilkd. 1909 ; 62 : 131.
[29] Liebmann JM, Tello C, Chew SJ, et al. Prevention of blinking alters iris configuration in pigment dispersion syndrome and in normal eyes. Ophthalmology. 1995 ; 102 : 446-55.
[30] Liu L, Ong EL, Crowston J. The concave iris in pigment dispersion syndrome. Ophthalmology. 2011 ; 118 : 66-70.
[31] Mastropasqua L, Ciancaglini M, Carpineto P, Gallenga PE. Early stadiation of pigmentary dispersion syndrome and long-term analysis of progression to pigmentary glaucoma. Am J Ophthalmol. 1996 ; 28 : 301-7.
[32] Migliazzo CV, Schaffer RN, Nykin R, Magee S. Long-term analysis of pigmentary dispersion syndrome and pigmentary glaucoma. Ophthalmology. 1986 ; 93 : 1528-36.
[33] Mora P, Sangermani C, Ghirardini S, et al. Ultrasound biomicroscopy and iris pigment dispersion: a case-control study. Br J Ophthalmol. 2010 ; 94 : 428-32.
[34] Moroi SE, Sieving PA, Nouri-Mahdavi K, et al. Long anterior zonules and pigment dispersion. Am J Ophthalmol. 2003 ; 136 : 1176-8.
[35] Murphy CG, Johnson M, Alvarado JA. Juxtacanalicular tissue in pigmentary and primary open angle glaucoma. The hydrodynamic role of pigment and others constituents. Arch Ophthalmol. 1992 ; 110 : 1779-85.
[36] Nagarajah S, Shun-Shin GA. Pigment deposition on the central aspect of the posterior lens capsule in pigmentary glaucoma. Digit J Ophthalmol. 2011 ; 17 : 69-71.
[37] Niyadurupola N, Broadway DC. Pigment dispersion syndrome and pigmentary glaucoma – a major review. Clin Exp Ophthalmol. 2008 ; 36 : 868-82.
[38] Pavlin CJ, Macken P, Trope G, et al. Ultrasound biomicroscopic features of pigmentary glaucoma. Can J Ophthalmol. 1994 ; 29 : 187-92.
[39] Potash SD, Tello C, Liebmann J, Ritch R. Ultrasound biomicroscopy in pigment dispersion syndrome. Ophthalmology. 1994 ; 101 : 332-9.
[40] Richardson TM, Hutchinson BT, Grant WM. The outflow tract in pigmentary glaucoma : a light and electron microscopic study. Arch Ophthalmol. 1977 ; 95 : 1015-25.
[41] Ritch R, Liebmann J, Robin A, et al. Argon laser trabeculolasty in pigmentary glaucoma. Ophthalmology. 1993 ; 100 : 909-13.
[42] Ritch R, Steinberger D, Liebmann JM. Prevalence of pigment dispersion syndrome in a population undergoing glaucoma screening. Am J Ophthalmol. 1993 ; 115 : 707-10.
[43] Scheie HG, Cameron JD. Pigment dispersion syndrome : a clinical study. Br J Ophthalmol. 1981 ; 65 : 264-9.
[44] Scheie HG, Fleischhauer HW. Idiopathic atrophy of the epithelial layers if the iris and ciliary body. Arch Ophthalmol. 1958 ; 59 : 216-28
[45] Scheie HG. Width and pigmentation of the angle of the anterior chamber ; a system of grading by gonioscopy. AMA Arch Ophthalmol. 1957 ; 58 : 510-2.
[46] Scott A, Kotecha A, Bunce C, et al. YAG laser peripheral iridotomy for the prevention of pigment dispersion glaucoma a prospective, randomized, controlled trial. Ophthalmology. 2011 ; 118 : 468-73.
[47] Scuderi G, Papale A, Nucci C, Cerulli L. Retinal involvement in pigment dispersion syndrome. Int Ophthalmol. 1996 ; 19 : 375-8.
[48] Sellem E. La trabéculoplastie a-t-elle encore des indications ? J Fr Ophtalmol. 2001 ; 24 : 1100-2.
[49] Siddiqui Y, Ten Hulzen RD, Cameron JD, et al. What is the risk of developing pigmentary glaucoma from pigment dispersion syndrome ? Am J Ophthalmol. 2003 ; 135 : 794-9.
[50] Sugar HS, Barbour FA. Pigmentary glaucoma : a rare clinical entity. Am J Ophthalmol. 1949 ; 32 : 90-2.
[51] Sugar HS. Pigmentary glaucoma : a 25-year review. Am J Ophthalmol. 1966 ; 62 : 499-507.
[52] Von Hippel E. Zur pathologischen anatomie des glaucoma. Arch Ophthalmol. 1901 ; 52 : 498.
[53] Weseley P, Liebmann J, Walsh JB, Ritch R. Lattice degeneration of the retina and the pigment dispersion syndrome. Am J Ophthalmol. 1992 ; 114 : 539-43.
[54] Wise JB, Witter SL. Argon laser therapy for open angle glaucoma, a pilot study. Arch Ophthalmol. 1979 ; 97 : 319-22.
[55] Yang JW, Sakiyalak D, Krupin T. Pigmentary glaucoma. J Glaucoma. 2001 ; 10 : 30-2.
[56] Zentmayer W. Association of an annular band of pigment on the posterior capsule of the lens with a Krukenberg spindle. Arch Ophthalmol. 1938 ; 20 : 52-7.
D. Gruber
Le syndrome exfoliatif (SXF) est une affection systémique diffuse de la matrice extracellulaire.
Le SXF s’observe partout dans le monde mais avec une prévalence très variable selon les régions. C’est la plus fréquente des causes identifiées de glaucome à angle ouvert. Il reste globalement sous-diagnostiqué.
Le gène de l’enzyme LOXL1 constitue un facteur de risque majeur pour le SXF et le glaucome exfoliatif (GXF), mais il est insuffisant pour provoquer la maladie.
La nature exacte du matériel exfoliatif (MXF) reste inconnue. Stress oxydatif élevé, TGF-β1 en excès et défenses antioxydantes diminuées sont des facteurs déterminants dans la complexe physiopathologie du SXF. La théorie pathogénique actuelle est une synthèse de la théorie de la membrane basale et de celle des fibres élastiques.
Le syndrome pré-exfoliatif permet de suspecter cliniquement la maladie. Le classique SXF ne se résume pas aux dépôts givrés sur la cristalloïde : kératopathie spécifique, vasculopathie irienne, fragilité zonulaire et, surtout, désorganisation progressive du trabéculum et déformabilité anormale de la lame criblée, ainsi que rupture de la barrière hémato-oculaire complètent l’atteinte intra-oculaire. Les preuves de manifestations extra-oculaires liées au SXF s’accumulent.
SXF et GXF sont souvent cliniquement unilatéraux. Le GXF présente une pression intra-oculaire (PIO) moyenne et des fluctuations tonométriques plus élevées que le glaucome primitif à angle ouvert (GPAO). La réponse au traitement médical est souvent insuffisante, justifiant le recours fréquent à la chirurgie.
Les perspectives thérapeutiques du glaucome secondaire que constitue le GXF sont celles des moyens susceptibles de stopper la formation et/ou la polymérisation du MXF ou de permettre sa dépolymérisation une fois constitué.
Le SXF est une affection systémique diffuse de la matrice extracellulaire. Il s’agit d’une microfibrillopathie caractérisée par la production excessive et l’accumulation progressive dans les tissus intra- et extra-oculaires d’un matériel fibrillaire extracellulaire anormal insoluble.
Le SXF prédispose à de multiples complications oculaires dont la principale est le glaucome. Il constitue la plus fréquente des causes identifiées de glaucome à angle ouvert.
Sur la base de données histopathologiques, il est maintenant clairement établi que le glaucome exfoliatif est une forme originale et spécifique de glaucome secondaire. Comme dans tout glaucome secondaire, c’est par le biais de l’hypertonie oculaire induite (en l’espèce par une « trabéculopathie exfoliative ») que se constitue principalement l’atteinte glaucomateuse.
Le SXF s’observe partout dans le monde mais avec une prévalence éminemment variable. Celle-ci est estimée en moyenne entre 10 et 20 % de la population générale de plus de 60 ans [16]. En France, Colin et al. [3] ont observé une prévalence de 20,6 % à Brest contre 3,6 % à Toulon chez des patients de 70 ans et plus. Chez les patients glaucomateux, la prévalence du SXF est élevée : de 10 à 30 % aux États-Unis, de 50 à 60 % en Europe du Nord. Les importantes différences observées selon les études découlent certes de vraies différences ethniques ou géographiques, mais aussi de la pertinence de l’examen diagnostique et de la population étudiée.
De multiples preuves d’hérédité ont été apportées par des études d’associations familiales mais aucun mode de transmission clair n’a été identifié.
L’association du gène de l’enzyme LOXL1 et du SXF/GXF a été établie en 2007 par Thorleifsson et al. [25] lors d’une étude princeps portant sur une population islandaise et suédoise. L’enzyme LOXL1 appartient à la superfamille « LOX » des enzymes extracellulaires. Elle est impliquée dans la synthèse et le métabolisme des fibres élastiques. Ce gène est situé sur le chromosome 15 (15q24). Deux polymorphismes nucléotidiques simples (SNP) ont été identifiés : rs 3825942 et rs 1048661. Les études dans d’autres pays ont confirmé ces résultats avec une constance pour le SNP rs 3825942 et des variations pour SNP rs 1048661. Le variant rs 3825942 a une prévalence très élevée, de 95 à 100 % chez les patients atteints. Toutefois, cette sensibilité très élevée est associée à une spécificité très faible car la prévalence est également grande chez les sujets sains (57 à 88 % selon les populations étudiées). En corollaire, un tel test génétique pour dépister les sujets à risque de SXF n’est pas pertinent. Il en est de même pour dépister les SXF à risque de GXF car la prévalence des SNP est sensiblement identique entre SXF et GXF [2].
Ce gène constitue un facteur de risque génétique majeur pour le SXF/GXF. Ce lien passe probablement par une réduction de l’expression de l’enzyme du gène de LOXL1 et de l’activité de l’enzyme. Cependant, ce gène n’est pas suffisant pour provoquer la maladie. Il représente une forte prédisposition mais des cofacteurs sont nécessaires. De plus, la prévalence également élevée chez les sujets sains suggère l’existence de gènes protecteurs possibles et/ou de facteurs environnementaux pouvant retarder l’apparition du SXF ou du GXF.
Le MXF est constitué de deux types de fibrilles immergées au sein d’une substance amorphe. On distingue les fibrilles de type A de 18-25 nm de diamètre, 1 µm de long avec des striations tous les 50 nm (fig. 14-7a,b) et les fibrilles de type B plus courtes (0,5 µm) et plus épaisses (30-40 nm). Ces fibrilles sont elles-mêmes composées par l’agrégation de microfibrilles de 5-10 nm de diamètre avec des striations de 10-12 nm entourées d’un matériel dense aux électrons, siège d’excroissances régulières responsables de la striation des fibrilles [17]. Ces caractéristiques sont suffisamment spécifiques en microscopie électronique pour distinguer clairement ce MXF de tout autre produit de la matrice extracellulaire
La composition chimique exacte du MXF reste inconnue. Il s’agit d’une structure complexe (constituée de glycoprotéines et de protéoglycanes) avec un noyau central protéique entouré d’une couronne glucidique (glycosaminoglycanes). Parmi les composants glucidiques (hydrocarbonés), outre les héparane-, chondroïtine-, dermatane-sulfates et l’acide hyaluronique, on retiendra le HNK-1 épitope impliqué dans l’adhérence du matériel exfoliatif sur les surfaces intra-oculaires [17]. La fraction protéique comprend des composants non collagènes des membranes basales (comme la laminine, la fibronectine) et des épitopes du système des fibres élastiques dont la fibrilline-1, constituant principal des microfibrilles élastiques et qui est un marqueur du MXF en microscopie immuno-électronique (fig. 14-7c).
L’origine du MXF est multifocale. Il est produit par une grande variété de types cellulaires intra- et extra-oculaires (cellules épithéliales, endothéliales, musculaires, du tissu conjonctif et de la paroi vasculaire) (fig. 14-7c,d et 14-8a,b). Il s’agit d’un processus biologique systémique actif, d’une fibrillogenèse qui s’accompagne progressivement d’une désorganisation de la membrane basale des cellules concernées pouvant aboutir à leur dégénérescence [18].

Fig. 14-7 Microscopie électronique à transmission montrant la structure et l’origine du MXF. a. Ultrastructure des fibrilles de MXF. b. Agrégation des microfibrilles (flèches) en fibrilles de MXF matures avec des striations de 50 nm (pointes de flèche). c. Immunomarquage à l’or colloïdal avec des anticorps antifibrilline-1 montrant clairement l’association du marqueur avec les fibrilles de MXF (PEX) émergeant d’une cellule de l’épithélium non pigmenté du corps ciliaire (NPE). d. Apparente production (flèches) de fibrilles de MXF (PEX) par une cellule endothéliale du trabéculum. (D’après Schlötzer-Schrehardt et Naumann [20]. Reproduction autorisée.)
Plusieurs théories pathogéniques ont été avancées. Elles s’appliquent à tous les tissus aussi bien extra-oculaires qu’oculaires. Si la théorie amyloïde a été définitivement écartée, deux théories restent d’actualité. Elles constituent probablement deux facettes du processus pathogénique.
Le MXF serait le produit d’un dysfonctionnement du métabolisme des membranes basales. Cette théorie est fondée sur la présence fréquente de MXF au contact de membranes basales de différentes sortes de cellules et sur des preuves immunohistochimiques de la présence d’épitopes des membranes basales au sein du MXF (laminine, héparane-sulfate, protéoglycane).
Le MXF serait le produit d’une dégénérescence particulière du tissu élastique, une élastose affectant les microfibrilles élastiques [17, 18]. Cette théorie s’appuie sur des similitudes structurelles entre le MXF et les fibres élastiques, et sur l’identification en immunohistochimie de fibrilline-1 au niveau du MXF, démontrant l’excessive production et l’agrégation anormale de fibres élastiques au sein du MXF (voir fig. 14-8a,b)
Elle pourrait être schématisée de la façon suivante [21] : en réponse à des facteurs déclenchants profibrotiques comme le stress oxydatif ou des facteurs de croissance (excès de TGF-β1 dans l’humeur aqueuse), le turnover de la matrice extracellulaire serait désorganisé (déséquilibre métalloprotéinases matricielles/inhibiteurs tissulaires des métalloprotéinases). L’enzyme LOXL1 hyperstimulée provoquerait un excès de cross-linking des composants élastiques de la matrice extracellulaire et des précurseurs du matériel exfoliatif. Il en résulterait, dans un environnement inflammatoire infraclinique mais chronique, aux défenses antioxydantes diminuées, une désorganisation spatiale des protéines qui échapperaient alors à toute protéolyse pour s’agréger en fibres anormales dans les tissus. Le syndrome exfoliatif pourrait ainsi être rattaché à la superfamille des « affections du repliement des protéines ».
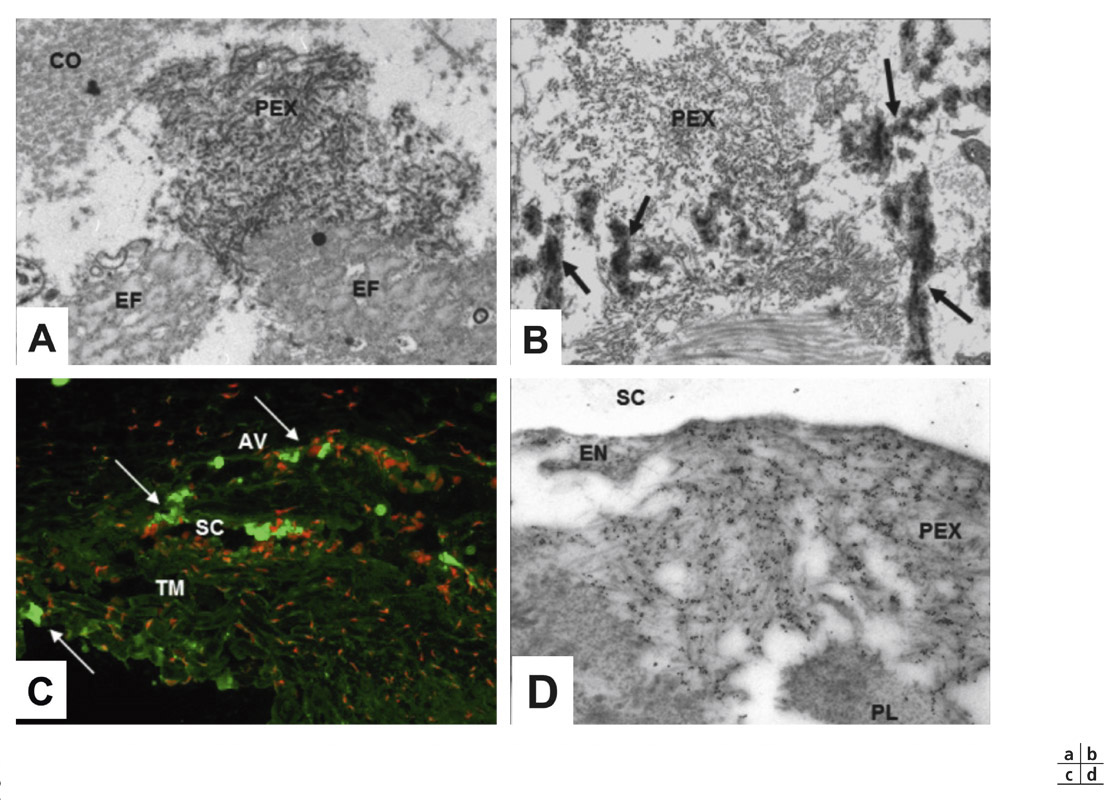
Fig. 14-8 Ultrastructure du MXF (a, b) et immunolocalisation de LOXL1 dans des dépôts de MXF dans des tissus intra-oculaires d’yeux de syndrome exfoliatif (c, d). a. Proximité immédiate entre des fibrilles de MXF (PEX) et des fibres élastiques (EF) dans du tissu conjonctif (CO : fibres de collagène). b. Proximité immédiate entre des fibrilles de MXF (PEX) et matériel élastique (flèches) dans la peau d’un patient atteint de syndrome exfoliatif. c. Immunolocalisation de LOXL1 (immunofluorescence verte et contre-coloration nucléaire en rouge) d’agrégats de MXF LOXL1-positifs (flèches) dans le réseau trabéculaire (TM) et à la périphérie du canal de Schlemm (SC) et de veines aqueuses (AV). d. Immunomarquage à l’or colloïdal de LOXL1 d’agrégats de MXF sous le mur interne de l’endothélium (EN) du canal de Schlemm (SC) à proximité immédiate de dépôts en plaques (PL). (D’après Schlötzer-Schrehardt [21]. Reproduction autorisée.)
Un examen attentif, bilatéral et comparatif est de rigueur pour le diagnostic du SXF qui reste avant tout clinique. SXF et GXF sont souvent cliniquement unilatéraux. En dix ans, environ un tiers des SXF se bilatéralisent et un tiers des SXF évoluent vers un glaucome. Par conséquent, nombreux sont les SXF qui resteront unilatéraux ou ne développeront jamais de glaucome.
Le classique SXF constitue un stade déjà avancé de la maladie. Il est précédé par un syndrome pré-exfoliatif moins connu, d’évolution lente et prolongée, et de diagnostic plus subtil. Son diagnostic clinique nécessite un examen attentif et orienté, sous dilatation pupillaire, en utilisant un faisceau étroit de la lampe à fente orienté à 45° et en comparant les deux yeux.
Au stade initial, un film mat homogène et diffus tapisse la capsule antérieure du cristallin. En s’épaississant, ce film est progressivement érodé par le frottement de l’iris, des défects localisés apparaissent puis s’élargissent et confluent pour réaliser finalement le classique tableau en cocarde [20].
Dès le stade précoce, d’autres signes de migration pigmentaire à partir de l’épithélium pigmenté de l’iris complètent le tableau [14]. Si aucun n’est spécifique (et même s’ils s’observent volontiers chez le sujet âgé), leur association est très évocatrice, surtout s’ils sont unilatéraux : irrégularité de la collerette pupillaire, pigment saupoudré sur le stroma irien péripupillaire, transillumination de l’iris péripupillaire, effet Tyndall pigmentaire lors de la dilatation pupillaire et surcharge pigmentaire irrégulière du trabéculum.
La détection en lumière infrarouge d’une transillumination irienne annulaire peut aider au diagnostic précoce.
Le MXF est présent dans l’ensemble de l’organisme. Il modifie non seulement toutes les structures du segment antérieur de l’œil mais aussi la plupart des tissus de l’organisme.
Les dépôts macroscopiques intra-oculaires de MXF sont visibles à la lampe à fente après dilatation pupillaire et examen attentif, bilatéral et comparatif. Le SXF intra-oculaire est souvent incomplet et/ou unilatéral. Cliniquement visible, il est complété par une atteinte histologique des autres structures de segment antérieur ainsi que des tissus extra-oculaires.
Il est le siège du signe le plus caractéristique, pathognomonique du SXF, avec des dépôts blanchâtres translucides d’aspect givré disposés en cocarde sur la cristalloïde : un disque central dans l’aire pupillaire, une couronne périphérique parfois enroulée sur elle-même avec des striations radiaires et une zone annulaire intermédiaire indemne en raison du balayage irien lors du jeu pupillaire (fig. 14-9). Ce tableau est parfois incomplet, avec absence du disque central dans 10 à 20 % des cas.
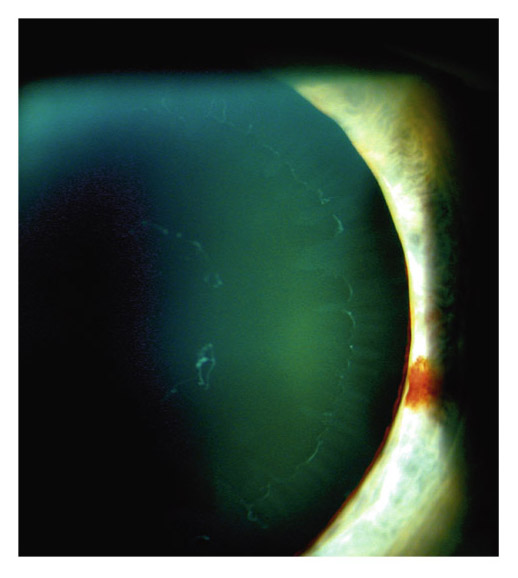
Fig. 14-9 Dépôts givrés de matériel exfoliatif disposés en cocarde sur la cristalloïde.
L’atteinte irienne est précoce et également caractéristique. Le MXF est présent sur la bordure pupillaire sous la forme de fins dépôts givrés irrégulièrement répartis, bien visibles au fort grossissement (fig. 14-10). De fins défects de l’épithélium pigmentaire irien sont responsables d’une transillumination de cette même collerette et/ou de l’iris juste adjacent, par opposition à la transillumination plus périphérique du syndrome de dispersion pigmentaire. Ce pigment arraché à l’épithélium irien par le frottement pupillaire sur le MXF cristallinien sous-jacent se disperse dans tout le segment antérieur, en particulier dans l’angle mais aussi sur le stroma irien où il s’observe disséminé uniformément lorsqu’il est abondant.

Fig. 14-10 Yeux pseudo-phakes, givre pupillaire (matériel exfoliatif) et atrophie de la collerette (œil droit, a), atrophie partielle de la collerette (œil gauche, b).
Elle est le siège d’une véritable kératopathie spécifique.
Des dépôts de MXF ainsi que des grains de pigment peuvent s’observer sur l’endothélium. Ils ne doivent pas être confondus avec des précipités inflammatoires. La densité cellulaire est diminuée aussi bien au niveau de l’épithélium basal que du stroma et de l’endothélium. La pachymétrie est plus élevée dans les yeux atteints. La densité des rameaux nerveux intracornéens est diminuée, constituant une neuropathie liée à la présence du MXF [30], expliquant la moindre sensibilité cornéenne. Il existe une production locale de MXF par les cellules endothéliales [4] associée à une phagocytose des grains de mélanine.
Le MXF visible sur les fibres zonulaires (fig. 14-11a) est également présent histologiquement au sein des fibres où il fragilise, voire interrompt, des fibres zonulaires [17], expliquant le phakodonesis éventuel et les complications possibles lors de la chirurgie de la cataracte. Il en est de même pour le déplacement vers l’avant du cristallin en procubitus qui favorise le bloc pupillaire et des fluctuations de la PIO.

Fig. 14-11 a. Vue gonioscopique : givre sur les fibres zonulaires (matériel exfoliatif) vu à travers l’iridectomie périphérique. b. Vue gonioscopique sur 6 heures d’un angle ouvert large avec hyperpigmentation régulière du trabéculum ainsi qu’en avant de l’anneau de Schwalbe (ligne de Sampaolesi).
Il est habituellement largement ouvert, avec une pigmentation marquée mais irrégulière qui déborde volontiers en avant de la ligne de Schwalbe, essentiellement au niveau de la portion inférieure de l’angle (ligne de Sampaolesi) (fig. 14-11b). Cette pigmentation tend à être plus marquée en cas de GXF comparé au SXF [5]. Une pigmentation grade 2 + ou 3 +, surtout en supérieur sur 12 heures, est très évocatrice de SXF [29].
L’association SXF-fermeture de l’angle n’est pas rare et ne doit pas être méconnue. Wishart et al. [29], sur une série de 76 patients avec syndrome exfoliatif, ont observé un angle étroit dans 32 % des cas : 18 % avec un angle susceptible de se fermer (angle closure suspect) et 14 % avec un angle fermé (angle closure), avec donc la présence de goniosynéchies.
Plusieurs facteurs propres au SXF prédisposent à la survenue d’une fermeture de l’angle en favorisant le blocage pupillaire : synéchies postérieures, rigidité d’où une moindre mobilité de l’iris, altération de la barrière hémato-aqueuse avec augmentation de la concentration en protéines de l’humeur aqueuse et bascule antérieure du cristallin, conséquence de l’hyperlaxité zonulaire [17].
Devant un angle susceptible de se fermer, la découverte d’un SXF constitue un argument supplémentaire pour une iridotomie périphérique préventive.
Le trabéculum est le siège, dans sa portion juxtacanaliculaire, de la principale zone de résistance à l’écoulement de l’humeur aqueuse, comme dans tout glaucome à angle ouvert. La gonioscopie ne permet d’apprécier que la pigmentation de l’angle. Celle-ci est habituellement marquée, quoique irrégulière. Elle ne traduit que la surcharge pigmentaire due à l’érosion de l’épithélium pigmenté postérieur de l’iris. Son importance est moindre que dans le syndrome de dispersion pigmentaire, mais l’essentiel n’est décelable qu’histologiquement puisqu’il s’agit de l’accumulation du MXF lui-même.
Le trabéculum est le siège d’une délétion de l’ADN mitochondrial liée au stress oxydatif, responsable d’une diminution des mitochondries à l’origine d’une perte prématurée des cellules trabéculaires [7]. Ce processus ne serait présent que dans le GXF et le GPAO à la différence des autres formes de glaucomes.
Le MXF présent au niveau du trabéculum a une double origine :
exotrabéculaire, apporté par le flux d’humeur aqueuse, responsable d’une obstruction « mécanique ». Ce facteur serait accessoire ;
intratrabéculaire, localement produit, qui serait le facteur principal [19].
Schlötzer-Schrehardt a montré que le MXF, après avoir été localement sécrété, s’accumule dans le tissu juxtacanaliculaire. Le processus débute au niveau des cellules endothéliales du canal de Schlemm, puis s’étend vers les portions plus internes pour englober progressivement tout le trabéculum, notamment les cellules endothéliales des canaux trabéculaires (voir fig. 14-7d ; fig. 14-8c,d). Il en résulte une désorganisation des deux barrières endothéliales ainsi que de la communication qui existe entre elles, et finalement un collapsus du canal de Schlemm. Contrairement au GPAO, on n’observe pas de dépôts en plaques ni de baisse de la cellularité trabéculaire.
Le GXF est donc bien un glaucome secondaire qui se distingue du glaucome primitif à angle ouvert non seulement cliniquement mais aussi histopathologiquement.
Comparé au GPAO, le GXF a une PIO moyenne et des pics tensionnels plus élevés ainsi que de plus grandes variations nycthémérales [6]. La PIO lors du diagnostic est souvent supérieure à 35 mmHg. La dilatation pupillaire peut déclencher un pic pressionnel par le biais de la libération pigmentaire induite.
Contrairement au GPAO, la réponse aux corticoïdes dans le GXF est comparable à celle des sujets normaux : seulement un tiers des patients est répondeur [17, 20]. De plus, il existe une corrélation entre la PIO et l’atteinte périmétrique lors du diagnostic, ainsi qu’entre les fluctuations tensionnelles diurnes et l’épaisseur de la couche des fibres nerveuses rétiniennes chez les patients avec GXF comparés aux GPAO. Cela suggère que l’atteinte glaucomateuse est plus directement pression-dépendante dans le GXF que dans le GPAO [20].
Il existe une rupture chronique de la barrière hémato-oculaire que pourrait confirmer le laser flare-cell meter. Elle est plus importante en cas de chirurgie que dans le GPAO, justifiant un traitement anti-inflammatoire postopératoire prolongé.
Le segment antérieur est le siège d’une ischémie bien objectivée en angiographie au vert d’indocyanine de l’iris, montrant hypoperfusion, micro-néovascularisation et vaisseaux anastomotiques. Cette vasculopathie irienne est liée à l’accumulation de MXF en position subendothéliale. Elle précède l’apparition histologique de MXF sur le corps ciliaire et le cristallin, et représente un stade préclinique précoce. Elle peut être la cause ou la conséquence de l’absence d’élimination de molécules anormales qui finissent par s’agréger sous forme de MXF. Enfin, elle explique la mauvaise dilatation pupillaire et la rigidité stromale.
Les synéchies postérieures sont plus fréquentes, même en l’absence de traitement myotique, en raison de la présence de MXF sur les deux surfaces (épithélium pigmenté et cristalloïde) qui aggrave le risque d’adhérence. Elles augmentent le blocage pupillaire et donc le risque de fermeture de l’angle [17].
La cataracte, en particulier nucléaire, apparaît plus fréquente en cas de SXF. Ritch et al. [17] ont émis l’hypothèse que le SXF favoriserait l’apparition d’une cataracte par le biais de l’ischémie oculaire qu’il entraîne. Surtout, le SXF augmente le risque de complications peropératoires en raison de la mauvaise dilatation pupillaire et de la fragilité zonulaire qui favorisent la rupture capsulaire. L’examen préopératoire se doit d’identifier ce facteur de risque que constitue un SXF évolué (phakodonesis, mauvaise dilatation pupillaire, rigidité irienne et hyperpigmentation trabéculaire) pour anticiper au mieux les difficultés opératoires. De plus, la fibrose du sac et la désintégration zonulaire progressive exposent à une luxation tardive du sac avec son implant dans le vitré.
La lame criblée et la sclère péripapillaire sont le siège d’une « élastinopathie » spécifique au SXF. Celle-ci est due à un déficit d’activité de LOXL1 [22] et à l’origine d’une déformabilité excessive, mesurable, de la lame criblée. Il en résulte une susceptibilité accrue de la tête du nerf optique aux agressions mécaniques et ischémiques en cas de glaucome.
La présence de MXF a été démontrée histologiquement non seulement dans la conjonctive mais aussi dans de nombreux tissus (peau, poumon, foie, rein, myocarde, méninges, vaisseaux) [23], signant une affection systémique diffuse même si son diagnostic clinique demeure actuellement uniquement ophtalmologique.
L’impact possible de la présence de MXF dans le développement et/ou l’exacerbation de pathologies systémiques reste pour l’essentiel à préciser [6]. Sur ce thème en pleine évolution, on retiendra une surdité neurosensorielle, une vasculopathie systémique et des marqueurs biologiques perturbés.
Elle est due à une atteinte de la cochlée et/ou du nerf auditif et des voies auditives centrales. Elle a été démontrée en comparant des SXF (sans glaucome) versus des témoins sains et des GXF versus des GPAO [12]. Les principales hypothèses pathogéniques évoquent la présence de MXF au niveau de l’organe de Corti et/ou au niveau de la paroi des vaisseaux de l’oreille interne et des voies auditives supérieures. Cette hypoacousie a une signification clinique car elle touche les fréquences importantes pour la compréhension de la parole, ce qui retentit sur la qualité de vie.
De même qu’il existe une relation démontrée entre la présence de MXF dans la paroi des vaisseaux iriens et la vasculopathie irienne (voir plus haut « Manifestations oculaires associées »), de nombreuses études ont également recherché les conséquences possibles in vivo des dépôts visibles dans la paroi des vaisseaux systémiques, gros vaisseaux et microcirculation.
Ainsi, il existe une fréquence significativement plus élevée des anévrismes de l’aorte abdominale sous-rénale chez les patients avec SXF ou GXF versus GPAO ou cataracte [4]. Un dépistage ciblé des anévrismes de l’aorte abdominale chez les patients à risque (SXF, homme, forte pigmentation de l’angle, diabète, HTA) apparaît justifié.
D’autres gros vaisseaux seraient également atteints. La rigidité de la carotide est augmentée, associée à une diminution de la sensibilité baroréflexe témoignant d’une altération du système de contrôle parasympathique cardiovasculaire. Cette pathologie augmente avec l’âge et le degré d’hyperhomocystéinémie. Il existe de surcroît un lien entre SXF et maladie vasculaire périphérique : l’index cheville/bras mesuré en Doppler couleur est significativement diminué chez les patients avec SXF de plus de 60 ans comparés aux témoins [13]. Enfin, la prévalence des hypersignaux dans la substance blanche en IRM suggère un lien entre ischémie cérébrale et SXF.
La microcirculation est altérée. Le flux sanguin capillaire sous-unguéal est diminué et le temps de réaction au test au froid est allongé comparativement à des GPAO et des sujets normaux du même âge. Ce résultat est indépendant d’un glaucome ou d’une hypertension intra-oculaire (HTO) associés éventuels. La fonction endothéliale est également perturbée. Comparés aux témoins, les SXF ont une moindre dilatation de l’artère brachiale induite aussi bien par l’hyperhémie (endothélium-dépendante) que par la nitroglycérine (endothélium-indépendante).
En revanche, le lien entre SXF et morbidité cardiovasculaire ou cérébrovasculaire reste fortement discuté [6].
L’association SXF et élévation de l’homocystéine plasmatique est bien démontrée [6]. L’hyperhomocystéinémie augmente le risque d’affections vasculaires (infarctus du myocarde, accident vasculaire cérébral ou thrombose veineuse). La balance oxydants/antioxydants est altérée dans l’humeur aqueuse et le sérum et les défenses antioxydantes sont diminuées [8]. Cela se traduit par un excès de radicaux libres responsables d’un stress oxydatif. Les taux de TGF-β1 et de CTGF (connective tissue growth factor) sont augmentés dans l’humeur aqueuse, ce qui favorise la surexpression et/ou la dérégulation de gènes impliqués dans la synthèse de la matrice extracellulaire [6].
Hyperhomocystéinémie, stress oxydatif augmenté et profil d’autoanticorps sériques particulier créent un syndrome inflammatoire infraclinique, jouant probablement un rôle dans la physiopathologie du SXF lui-même [21] (voir plus haut « Théorie pathogénique moléculaire actuelle »).
Lors du diagnostic d’un SXF, environ la moitié des patients ont une atteinte unilatérale, bien que l’atteinte soit probablement toujours histologiquement bilatérale. Le terme « cliniquement unilatéral » est à préférer à celui d’« unilatéral », trop imprécis et source de confusion.
Selon une étude prospective portant sur 63 patients, Puska [15] a observé 38 % de conversion vers une atteinte bilatérale en dix ans (méthode Kaplan-Meier). Tarkkanen et Kivelä [24], dans une étude rétrospective et avec la méthode d’analyse d’incidence cumulative, retrouvent un chiffre de 36 à 52 % à dix ans (selon le critère retenu pour fixer la date de conversion). Ces chiffres ne tiennent pas compte des atteintes précoces, avant les dépôts pathognomoniques sur le cristallin (voir plus haut « Syndrome pré-exfoliatif »). Des facteurs lors du diagnostic d’unilatéralité pourraient raccourcir le délai de conversion (selon la méthode d’analyse) : un âge avancé et/ou une faible différence de PIO entre les deux yeux [24]. Cela signifie qu’une majorité de patients n’auront pas de bilatéralisation de leur SXF de toute leur vie.
Les raisons de cette asymétrie fréquente et prolongée sont mal comprises. On évoque des facteurs locaux oculaires et/ou extra-oculaires comme une différence de production d’humeur aqueuse, de facilité d’écoulement ou de vascularisation oculaire [6] ou bien encore des facteurs génétiques ou immunologiques. Des sous-types de SXF (qui restent à définir) pourraient aussi être en cause.
Le taux de conversion SXF/GXF observé par Puska [15] est de 32 % à dix ans. Il est de 38 % pour l’œil adelphe sans SXF initial. Une PIO initiale élevée augmente le risque de conversion (risque relatif : 1,47). Après avoir sorti la PIO initiale du modèle, on observe deux autres facteurs prédictifs de conversion : une mauvaise dilatation pupillaire et une plus grande différence de PIO avec l’œil initialement sans SXF. Cette conversion survient rapidement : deux tiers dans les trois ans, 94 % dans les cinq ans.
Le taux de conversion vers un glaucome pour les HTO avec SXF est le double de celui des HTO sans SXF. Le SXF est un très important facteur de risque indépendant de conversion d’une HTO vers le glaucome.
Les raisons qui font que seulement certains SXF évoluent vers un GXF restent là encore mal comprises. La simple quantité de MXF et les différences interindividuelles dans la gestion des perturbations métaboliques ou des facteurs génétiques ont été évoquées.
Le glaucome est souvent diagnostiqué en même temps que le SXF ou peu après. Il survient parfois beaucoup plus tard (eCas clinique 14-1), voire à l’inverse il précède le SXF. Ce risque d’HTO et de GXF semble cumulatif dans le temps [15]. Il est donc de bonne pratique clinique de toujours rechercher attentivement un SXF chaque fois que l’on diagnostique un GPAO.
Glaucome exfoliatif suivi pendant 25 ans
1. Mr L., 65 ans, consulte en mai 1986 pour une hypertonie oculaire (HTO) connue et traitée. Ni antécédent familial de glaucome ni facteur de risque systémique ne sont relevés. 10/10 ODG. PIO : OD 28 mmHg, OG 13 mmHg avec traitement. La pachymétrie (qui sera mesurée en 2000) est un peu fine à 509 μm OD et 506 μm OG. L’angle iridocornéen est ouvert, large, avec une pigmentation antérieure en inférieur OD. Le syndrome exfoliatif est patent, unilatéral droit. La papille droite est manifestement glaucomateuse (fig. A). Le champ visuel (fig. B) est conforme à l’état papillaire OD et normal OG.
Attitude pratique : trabéculorétraction au laser à l’argon (TLA) OD seul.
2. La PIO remonte en 1992 à 30 mmHg avec traitement (fig. C).
Attitude pratique : trabéculectomie OD.
3. Baisse visuelle OD en 1996 : 5/10f : cataracte nucléaire. PIO : 16 mmHg.
Attitude pratique : phako- exérèse OD.
4. Dès la période postopératoire, la PIO remonte OD à 44 mmHg et même 50 mmHg, signant une décompensation de la fistule de trabéculectomie (fig. C)
Attitude pratique : sclérectomie profonde non perforante OD.
5. Une HTO OG apparaît en 1997, le syndrome exfoliatif ne devenant cliniquement visible OG qu’en janvier 1998 (fig. C, repère y)
Attitude pratique : trabéculorétraction au laser à l’argon OG.
6. Phako- exérèse OG en 2008.
7. Depuis et jusqu’en 2012 (date du décès) : poursuite d’une surveillance régulière, avec champ visuel semestriel, bithérapie OG seul. Au dernier contrôle, la vision est à 6/10f OD et 10/10 OG avec correction et la PIO : OD 11,5 mmHg sans traitement, OG 10,5 mmHg sous acétazolamide et cartéolol LP.
Commentaires
Le syndrome exfoliatif est une bonne indication de la trabéculorétraction au laser à l’argon même si son efficacité est limitée dans le temps.
Le gain tonométrique s’est maintenu chez ce patient pendant six ans à l’œil droit, quatre ans à l’œil gauche (fig. C, repère z), ce qui est conforme aux données de la littérature.
Le syndrome exfoliatif est souvent cliniquement unilatéral. Le décalage observé ici, d’au moins 12 ans entre l’atteinte clinique de l’œil droit et celle de l’œil gauche, est remarquable. La réalité d’une atteinte infraclinique est d’ailleurs suggérée par l’évolution tonométrique de l’OG : une HTO apparaît (avril 1996, voire mai 1993, fig. C, repère x), soit bien avant l’apparition clinique des dépôts précristalliniens (janvier 1998, fig. C, repère y).
La décompensation de la fistule d’une trabéculectomie efficace par l’intervention de cataracte est malheureusement classique. Le patient doit être prévenu de ce risque.
Un suivi rigoureux, avec une pression cible effectivement atteinte (PIO ≤ 12 mmHg à tous les contrôles depuis septembre 1998) permet de stabiliser le champ visuel sur le long terme (fig. B et D) avec une détérioration du MD de 0,1 dB/an, ce qui se rapproche de l’évolution des sujets normaux avec l’âge.

Fig. A OD : excavation papillaire avec amincissement du bord neurorétinien en inférieur et pâleur. OG : bord neurorétinien régulier, respect de la règle ISNT (Mr L., 2002).

Fig. B Champs visuels Octopus G2 dynamique. OD : déficits peu évolutifs entre 1998 et 2011. OG : reste dans les limites de la normale.

Fig. C Courbe de tension oculaire de 1986 à 2011. La PIO de l’OD se maintient en dessous de 15 mmHg sans traitement depuis la sclérectomie profonde de 1997. La pression cible (18 mmHg ?) est également atteinte pour l’OG avec une bithérapie.
Fig. D Champs visuels : analyse de tendance et clusters. OD entre 1997 et 2011 (depuis la sclérectomie) : pente du MD de 0,1 dB/an, ce qui se rapproche de la perte normale avec l’âge mais défects localisés significatifs, confirmés par l’analyse en cluster en temporal inférieur et surtout maculaire supérieur. OG entre 1995 et 2011 : stabilité avec pente du MD de 0 dB/an.
Le diagnostic est fait en moyenne à un stade plus évolué et la réponse au traitement est plus faible que dans le GPAO, expliquant le recours fréquent à un traitement chirurgical.
Plusieurs facteurs de comorbidité compliquent la prise en charge des GXF : les patients sont en moyenne plus âgés, la cataracte est plus fréquente, et la fragilité zonulaire comme la mauvaise dilatation pupillaire augmentent le risque chirurgical.
Dans le GXF, le principal facteur de risque est la PIO. Surtout, le taux de progression du champ visuel dans le GXF en l’absence de traitement est le plus élevé des glaucomes chroniques à angle ouvert. Heijl et al. [5], en reprenant les patients de l’étude EMGT, ont observé que le taux moyen de dégradation du MD du champ visuel était de 3,13 dB/an dans le GXF comparé à 1,31 dB/an pour le GPAO avec HTO et 0,36 dB/an pour le glaucome à pression normale. Cette progression est significativement plus rapide pour le sous-groupe de patients âgés de plus de 68 ans : 1,48 dB/an versus 0,6 dB/an pour les plus jeunes, et l’on connaît la fréquence accrue du GXF avec l’âge. Enfin, durant les six ans de l’étude, une progression du champ visuel a été rapportée respectivement pour 93 % des GXF, 74 % des GPAO avec HTO et 56 % des glaucomes à pression normale.
Le GXF progresse rapidement et le SXF constitue en lui-même un important facteur de risque de progression indépendant de la PIO.
Comme dans tout glaucome, le principal moyen thérapeutique reste l’abaissement de la PIO. Les moyens sont comparables à ceux utilisés pour le GPAO. Certaines particularités méritent toutefois d’être analysées.
Le GXF est réputé dans la littérature répondre moins bien au traitement médical que le GPAO. Il s’agirait plutôt, d’après Hollo et Konstas [6], d’un statut tonométrique plus sévère que d’un manque d’efficacité en soi. Le gain tonométrique direct ainsi que le pourcentage de baisse de la PIO sous monothérapie est même plus élevé que pour des GPAO appariés. En revanche, les PIO restent plus élevées et la pression cible est plus rarement atteinte.
Il existe une relation directe entre la réduction de la PIO et le taux de progression. Le GXF est « PIO-dépendant ». Dans une étude rétrospective portant sur 167 patients avec GXF suivis cinq ans, Konstas et al. [9] ont observé que 28 % des patients avec une PIO moyenne inférieure ou égale à 7 mmHg ont un champ visuel qui s’aggrave, comparés à 70 % pour le groupe ayant une PIO moyenne supérieure ou égale à 20 mmHg.
La pression cible étant difficile à atteindre, le recours à des associations médicamenteuses s’impose souvent d’emblée, avec ou sans combinaisons fixes.
Au-delà de leur efficacité tonométrique connue, trois classes thérapeutiques ont (ou auraient) un impact spécifique dans le GXF :
les inhibiteurs de l’anhydrase carbonique (IAC) présentent un inconvénient théorique car, en diminuant le flux à travers le trabéculum, ils favorisent l’accumulation du MXF et du pigment, ce qui aggraverait la fonction trabéculaire. La PIO après arrêt des IAC remonterait au-dessus de la PIO initiale [17] ;
les myotiques limitent le frottement iridocristallinien et donc la surcharge pigmentaire du trabéculum. Ils favorisent également le flux trabéculaire. À l’inverse, ils favorisent la création de synéchies iridocristalliniennes qui augmentent le risque de blocage pupillaire. Ils aggravent l’impact visuel d’une cataracte éventuellement associée ;
le latanoprost diminue la concentration dans l’humeur aqueuse du TGF-β1 actif, des métalloprotéinases matricielles (MMP2) et des inhibiteurs tissulaires des métalloprotéinases (TIMP2), qui sont autant de paramètres impliqués dans la physiopathologie du MXF [10]. Il reste à préciser s’il s’agit d’un effet de classe ou propre au latanoprost et surtout si cela peut réduire l’accumulation de MXF en cas d’utilisation précoce.
La plupart des études s’accordent pour attribuer à la trabéculoplastie au laser à l’argon (TLA) un effet initial meilleur dans le GXF comparé au GPAO [26]. En moyenne, le gain tonométrique avoisine les 30 %. En revanche, l’efficacité diminue régulièrement avec le temps, ce qui n’est pas surprenant puisque le processus biologique se poursuit au niveau du trabéculum avec surcharge en MXF et pigment.
La trabéculoplastie au laser sélectif (SLT) dans le GXF a une efficacité et une sécurité équivalente à la TLA (comme dans le GPAO).
Compte tenu de sa bonne tolérance, le SLT peut être envisagé (comme pour la TLA) aussi bien en 1re intention que pour éviter une escalade médicamenteuse (ou réduire un traitement mal toléré) ou enfin pour retarder une chirurgie. Dans tous les cas, il faut prévenir tout pic tensionnel post-laser immédiat (apraclonide avant et juste après la procédure) et assurer un traitement anti-inflammatoire pendant 8 à 15 jours. L’avantage de pouvoir renouveler ce traitement doit encore être confirmé dans cette forme clinique de glaucome secondaire à angle ouvert.
L’iridectomie périphérique a toute sa place en cas de fermeture de l’angle associée au SXF.
Le recours à la chirurgie est fréquent dans le GXF [6, 17]. La trabéculectomie reste la chirurgie de référence pour le GXF. Sa technique et ses résultats ne sont pas différents de ceux du GPAO. Les indications de chirurgie combinée cataracte-glaucome sont fréquentes compte tenu de l’âge des patients.
D’autres techniques ont fait la preuve de leur efficacité :
la sclérectomie profonde non perforante a une efficacité tonométrique équivalente dans le GXF comparé au GPAO. Elle aurait l’avantage par rapport à la trabéculectomie de limiter l’inflammation postopératoire ainsi que le risque d’hypotonie et de cataracte [11] ;
la viscocanalostomie a fait la preuve de son efficacité dans une étude prospective sur cinq ans (GPAO et quelques GXF). Une étude prospective comparant combinée phaco-viscocanolostomie chez des GXF versus GPAO a conclu à une réduction moyenne de PIO plus grande dans le groupe GXF [1] ;
la trabéculo-aspiration ou trabéculectomie ab interno est une nouvelle technique qui vise à augmenter le flux trabéculaire en aspirant les débris trabéculaires et le pigment de l’angle. Ting et al. [27], dans une étude prospective, ont comparé l’efficacité de la trabéculectomie ab interno dans le GXF versus GPAO (avec ou sans phaco-). Le gain tonométrique moyen à un an est meilleur dans le groupe GXF.
La rupture de la barrière hémato-acqueuse déjà présente spontanément est aggravée par toutes ces chirurgies et justifie un traitement anti-inflammatoire postopératoire rigoureux et prolongé.
Le GXF étant un glaucome secondaire, les perspectives portent sur le développement des moyens susceptibles de stopper la formation ou la polymérisation du MXF ou de permettre sa dépolymérisation une fois constitué. Les molécules clés de la cascade physiopathologique du MXF sont autant de cibles thérapeutiques spécifiques potentielles (TGF-β1, TIMP, CTGF). La réduction du stress oxydatif, de l’homocystéinémie, de l’expression de l’enzyme LOXL1 dans les tissus atteints et la modification des récepteurs de l’adénosine A3 sont également des voies prometteuses. Enfin, la découverte récente d’un modèle animal proche du SXF chez une souris ouvre de nouvelles voies de recherche [28].
Retenir
Le SXF constitue la plus fréquente des causes identifiées de glaucome à angle ouvert. Son diagnostic passe par un examen attentif bilatéral et comparatif.
L’association du gène de l’enzyme LOXL1 et du SXF/GXF est établie. Cependant, un test génétique de dépistage, fondé sur l’identification des SNP correspondants, n’est pas possible car la prévalence chez les sujets sains est trop élevée.
Le GXF est un glaucome secondaire qui se distingue génétiquement mais aussi cliniquement et physiopathologiquement du glaucome primitif à angle ouvert (GPAO). L’association SXF-glaucome chronique par fermeture de l’angle n’est pas rare. Elle est sous-estimée. Il est de bonne pratique clinique de rechercher minutieusement un SXF devant tout glaucome, qu’il soit à angle ouvert ou par fermeture de l’angle.
L’identification des manifestations systémiques du SXF est en plein développement : surdité neurosensorielle, vasculopathie des gros vaisseaux mais aussi de la microcirculation semblent établies. Le patient doit en être informé.
Le pronostic du GXF est plus sévère que celui du GPAO. Traitements et suivis se doivent d’être particulièrement rigoureux.
Mauvaise dilatation pupillaire et fragilité zonulaire augmentent les risques de complications lors de la chirurgie de la cataracte en cas de SXF. L’examen préopératoire se doit d’identifier les yeux à risque pour une anticipation optimale.
Le recours à la chirurgie est fréquent dans le GXF et la trabéculectomie reste sa chirurgie de référence.
[1] Awadalla MA, Hassan KM. Phacoviscocanalostomy in pseudoexfoliation glaucoma versus primary open-angle glaucoma. Can J Ophthalmol. 2011 ; 46 : 77-82.
[2] Challa P. Genetics of pseudoexfoliation syndrome. Curr Opin Ophthalmol. 2009 ; 20 : 88-91.
[3] Colin J, Le Gall G, Le Jeune B, Cambrai MD. The prevalence of exfoliation syndrome in different areas of France. Acta Ophthalmol Suppl. 1988 ; 184 : 86-9.
[4] Djordjevic-Jocic J, Jovanovic P, Bozic M, et al. Prevalence and early detection of abdominal aortic aneurysm in pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Curr Eye Res. 2012 ; 37 : 617-23.
[5] Heijl A, Bengtsson B, Hyman L, et al. Natural history of open-angle glaucoma. Ophthalmology. 2009 ; 116 : 2271-6.
[6] Hollo G, Konstas AGP. Exfoliation syndrome and exfoliative glaucoma. 2nd ed. Savona, Dogma Srl, 2012.
[7] Izzotti A, Longobardi M, Cartiglia C, et al. Mitochondrial damage in the trabecular meshwork occurs only in primary open-angle glaucoma and in pseudoexfoliation glaucoma. PlosOne. 2011 ; 6 : 1-7.
[8] Koliakos GG, Befani CD, Mikropoulos D, et al. Prooxidant-antioxidant balance, peroxide and catalase activity in the aqueous humour and serum of patients with exfoliation syndrome or exfoliative glaucoma. Graefe’s Arch Clin Exp Ophthalmol. 2008 ; 246 : 1477-83.
[9] Konstas AGP, Hollo G, Astakhov YS, et al. Factors associated with long-term progression or stability in exfoliation glaucoma. Arch Ophthalmol. 2004 ; 122 : 29-33.
[10] Konstas AGP, Koliakos G, Karabatsas CH, et al. Latanoprost therapy reduces the levels of TGF beta 1 and gelatinases in the aqueous humour of patients with exfoliative glaucoma. Exp Eye Res. 2006 ; 82 : 319-22.
[11] Ollikainen ML, Puustjärvi TJ, Rekonen PK, et al. Mitomycin C-augmented deep sclerectomie in primary open-angle glaucoma and exfoliation glaucoma : a three-year prospective study. Acta Ophthalmol. 2011 ; 89 : 548-55.
[12] Paliobei VP, Psillas GK, Mikropoulos DG, et al. Hearing evaluation in patients with exfoliative and primary open-angle glaucoma. Otolaryngol Head Neck Surg. 2011 ; 145 : 125-30.
[13] Praveen MR, Shah SK, Vasavada AR, et al. Pseudoexfoliation as a risk factor for peripheral vascular disease : a case-control study. Eye. 2011 ; 25 : 174-9.
[14] Prince AM, Streeten BW, Ritch R, et al. Preclinical diagnosis of pseudoexfoliation syndrome. Arch Ophthalmol. 1987 ; 105 : 1076-82.
[15] Puska P. Unilateral exfoliation syndrome : conversion to bilateral exfoliation and to glaucoma : a prospective 10-year follow-up study. J Glaucoma. 2002 ; 11 : 517-24.
[16] Ringvold A. Epidemiology of the pseudo-exfoliation syndrome. Acta Ophthalmol Scand. 1999 ; 77 : 371-5.
[17] Ritch R, Schlötzer-Schrehardt U. Exfoliation syndrome. Surv Ophthalmol. 2001 ; 45 : 265-315.
[18] Ritch R, Schlötzer-Schrehardt U, Konstas AG. Why is glaucoma associated with exfoliation syndrome ? Progress in Retinal and Eye Research. 2003 ; 22 : 253-75.
[19] Schlötzer-Schrehardt U, Naumann GOH. Trabecular meshwork in pseudoexfoliation syndrome with and without open-angle glaucoma. Invest Ophthalmol Vis Sci. 1995 ; 36 : 1750-64.
[20] Schlötzer-Schrehardt U, Naumann GOH. Pseudoexfoliation glaucoma. In : Kriegelstein GK, Weinreb RN (eds). Essentials in ophthalmology-glaucoma. Heidelberg, Springer, 2004 : 157-76.
[21] Schlötzer-Schrehardt U. Molecular pathology of pseudoexfoliation syndrome/glaucoma. New insights from LOXL1 gene associations. Exp Eye Res. 2009 ; 88 : 776-85.
[22] Schlötzer-Schrehardt U, Hammer CM, Krysta AW, et al. LOXL1 deficiency in the lamina cribrosa as candidate susceptibility factor for a pseudoexfoliation-specific risk of glaucoma. Ophthalmology. 2012 ; 119 : 1832-43.
[23] Streeten BW, Li ZY, Wallace RN, et al. Pseudoexfoliative fibrillopathy in visceral organs of a patient with pseudoexfoliation syndrome. Arch ophthalmol. 1992 ; 110 : 1757-62.
[24] Tarkkanen A, Kivelä T. Cumulative incidence of converting from clinically unilateral to bilateral exfoliation syndrome. J Glaucoma. 2004 ; 13 : 181-4.
[25] Thorliefsson G, Magnusson KP, Sulem P, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science. 2007 ; 317 : 1397-1400.
[26] Threlkeld AB, Hertzmark E, Sturm RT, et al. Comparative study of the efficacy of argon laser trabeculoplasty for exfoliation and primary open-angle glaucoma. J Glaucoma. 1996 ; 5 : 311-6.
[27] Ting JL, Damji KF, Stiles MC. Ab interno trabeculectomy : outcomes in exfoliation versus primary open-angle glaucoma. J Cataract Refract Surg. 2012 ; 38 : 315-23.
[28] Trantow CM, Mao M, Petersen GE, et al. Lyst mutation in mice recapitulates iris defects of human exfoliation syndrome. Invest Ophthalmol Vis Sci. 2009 ; 50 : 1205-14.
[29] Wishart PK, Spaeth GL, Poryzees EM. Anterior chamber angle in the exfoliation syndrome. Br J Ophthalmol 1985 ; 69 : 103-5.
[30] Zheng X, Shiraishi A, Okuma S, et al. In vivo confocal microscopic evidence of keratopathy in patients with pseudoexfoliation syndrome. Invest Ophthalmol Vis Sci. 2011 ; 52 : 1755-61.
M. Poli, E. Sellem
Le glaucome éteint traduit l’extinction d’un glaucome précédemment évolutif, avec de nombreux mécanismes répertoriés :
augmentation transitoire de la résistance à l’évacuation de l’humeur aqueuse : trabéculaire (glaucomes pigmentaire, cortisonique, uvéitique, traumatique), uvéosclérale par élévation de la pression veineuse épisclérale (orbitopathie dysthyroïdienne, malformations artérioveineuses, fistule, etc.) ;
fermeture transitoire de l’angle iridocornéen de cause :
cristallinienne : cataracte (fermeture primitive de l’angle), luxation antérieure du cristallin,
ciliaire : kystes ciliaires, œdème inflammatoire du corps ciliaire,
choroïdienne : turgescence, effusion, décollement ou tumeur choroïdienne,
sclérale : iatrogénique par indentation sclérale ;
hypersécrétion transitoire d’humeur aqueuse (crises glaucomatocyclitiques) ;
atrophie secondaire du corps ciliaire.
Sa prise en charge spécifique doit être bien connue.
Les glaucomes éteints (burned-out ou burnt-out glaucoma des Anglo-Saxons) sont une entité clinique caractérisée par une neuropathie optique glaucomateuse associée à une perte caractéristique du champ visuel, non évolutive et en l’absence d’hypertension intra-oculaire (HTO). Ils traduisent l’extinction d’un glaucome précédemment évolutif avec habituellement une élévation transitoire, mais dommageable, de la pression intra-oculaire (PIO) : HTO à angle ouvert (secondaire ou, très controversée, primitive) ou par fermeture de l’angle dont le facteur causal a été soustrait.
Sont exclus de cette définition les glaucomes dont la progression a été stoppée à l’instauration d’un traitement hypotonisant médical, physique et/ou chirurgical.
Les résultats de la Collaborative Normal-Tension Glaucoma Study (CNTGS) indiquent que 65 % des glaucomes à pression normale (GPN) ne présentent pas d’évolution clinique après cinq ans de suivi. Ces GPN représentent donc une véritable forme frontière avec les glaucomes éteints, dont la prévalence est difficile à évaluer du fait de l’existence de progresseurs lents, qui peuvent prendre plus de temps à se révéler [14].
Les glaucomes éteints sont souvent l’apanage des personnes âgées [19]. Il peut s’agir de formes unilatérales isolées ou bilatérales. Ils peuvent être mis en évidence de façon fortuite, parfois des décennies après la constitution de l’atteinte glaucomateuse, ou lors du suivi d’une HTO ancienne connue et résolutive.
L’examen du fond d’œil découvre une papille typiquement glaucomateuse, dont l’excavation est souvent importante [19, 39]. Lors du suivi, elle ne présentera toutefois aucun signe d’évolutivité (augmentation de l’excavation ou hémorragie).
L’observation directe du segment antérieur et la gonioscopie s’attacheront à mettre en évidence d’éventuels signes cliniques témoignant d’un mécanisme à l’origine d’une HTO ancienne : dispersion pigmentaire, atrophie irienne périphérique, séquelles d’inflammation intra-oculaire ou de traumatisme oculaire, dépôts pigmentés d’accolement iridocornéen.
La PIO est considérée comme normale dans les glaucomes éteints, soit inférieure à 21 mmHg, en s’assurant par une pachymétrie que l’épaisseur de la cornée centrale n’est pas anormalement basse. Éventuellement, la réalisation d’une courbe de la PIO peut permettre d’éliminer l’existence de pics pressionnels.
L’examen du champ visuel met en évidence des déficits glaucomateux caractéristiques. Au cours du suivi, il ne sera pas mis en évidence de progression de ces déficits. Toutefois, lorsque le glaucome est très évolué (îlot central, excavation totale), la perte physiologique des cellules ganglionnaires peut, sur le long terme, aggraver à elle seule la situation périmétrique et faire mettre en doute la non-progressivité du glaucome.
S’ils sont réalisés, ils confirment l’atteinte de la tête du nerf optique et la raréfaction des fibres nerveuses rétiniennes. Le suivi sur le long cours ne doit pas indiquer de progression.
Les glaucomes éteints sont la traduction de l’extinction d’un glaucome autrefois évolutif. Ils peuvent donc résulter :
d’une augmentation transitoire de la résistance à l’évacuation de l’humeur aqueuse, d’origine trabéculaire ou uvéosclérale ;
d’une fermeture transitoire de l’angle iridocornéen ;
d’une hypersécrétion transitoire d’humeur aqueuse ;
d’une atrophie secondaire du corps ciliaire abaissant la PIO.
La soustraction du phénomène responsable du déséquilibre de la balance production/évacuation de l’humeur aqueuse restitue une PIO normale, en laissant pour séquelle une neuropathie optique glaucomateuse devenue non progressive et associée à des déficits périmétriques caractéristiques.
La voie trabéculaire est la principale voie d’évacuation (80 %) de l’humeur aqueuse. Le réseau trabéculaire, composé de cellules et de matrice extracellulaire (MEC), régule le flux excrétoire d’humeur aqueuse et contrôle ainsi la PIO (voir chapitre 7) [20, 29].
Ainsi, une HTO peut être la conséquence :
d’un blocage trabéculaire : pigmentaire, cellulaire (hématies ou cellules inflammatoires), fibrineux ou composé d’autres débris ;
de modifications morphologiques et biochimiques du trabéculum par remodelage de la MEC trabéculaire ;
d’une réduction du calibre des pores trabéculaires par un œdème trabéculaire inflammatoire ;
d’un dysfonctionnement des cellules endothéliales trabéculaires.
La réversibilité de cette résistance à l’écoulement trabéculaire de l’humeur aqueuse s’explique par les propriétés phagocytaires propres du trabéculum [10] et/ou le remodelage permanent de la composition de la MEC du réseau trabéculaire par de multiples protéases [16, 22, 40].
Le glaucome pigmentaire (GP), glaucome secondaire à angle ouvert décrit en détail dans le chapitre 14-I, est lié à un encombrement trabéculaire par des granules pigmentaires issus d’un contact mécanique anormal entre les fibres de la zonule et l’épithélium irien, lui-même secondaire à un bloc pupillaire inverse [11, 12, 21, 34, 38]. L’élévation de la PIO est secondaire à un encrassement des espaces intertrabéculaires par ces granules pigmentaires et des débris cellulaires, à une raréfaction des cellules trabéculaires [17, 27], à une dégénérescence cellulaire macrophagique et endothéliale, à la fusion des lamelles trabéculaires associée au collapsus des espaces intertrabéculaires, à une augmentation de la MEC trabéculaire ainsi qu’à une oblitération du canal de Schlemm [17, 41].
Après une phase active de dispersion pigmentaire, une phase de régression peut être observée et parfois, avec elle, une normalisation spontanée de la PIO. Les déficits transilluminables iriens disparaissent, probablement par migration et/ou glissement des cellules épithéliales adjacentes, alors que diminuent les pigmentations irienne antérieure, endothéliales cornéenne et trabéculaire, prédominant à ce stade dans la portion supérieure de l’angle iridocornéen. En revanche, il persiste souvent des reliquats du fuseau de Krukenberg – s’il a existé – avec une granulation raréfiée et plus claire sur l’endothélium cornéen, ainsi que les dépôts pigmentaires rétro-iriens au niveau de l’équateur cristallinien (ligne de Scheie) et de la hyaloïde antérieure, permettant souvent d’affirmer la responsabilité ancienne de la dispersion pigmentaire. Enfin, chez ces sujets, en grande majorité des hommes myopes, l’hyperconcavité de l’iris périphérique à l’origine du contact iridozonulaire persiste à l’examen gonioscopique [25, 32].
Dans le cas du glaucome pigmentaire éteint, il est naturel que les patients soient souvent plus âgés que la moyenne des patients atteints de GP évolutifs [32] (eCas clinique 14-2).
Deux hypothèses physiopathologiques expliquent cette régression :
l’accroissement normal du volume cristallinien augmente le bloc pupillaire physiologique, réduisant le contact iridozonulaire en propulsant l’iris vers l’avant ;
les capacités d’accommodation diminuent, réduisant les frottements iridozonulaires [33].
Le remodelage de la MEC trabéculaire et la phagocytose des débris pigmentaires et cellulaires du réseau trabéculaire autorisent alors la réduction de la résistance à l’écoulement de l’humeur aqueuse et la normalisation de la PIO.
Glaucome éteint
Madame P. a 70 ans lorsqu’elle consulte en 2001 pour une baisse d’acuité visuelle à 3/10 non améliorable. Elle est myope alors de – 7,00 dioptries et a une cataracte nucléaire. Toutefois, la papille gauche présente une excavation importante et très suspecte (fig. A, après phaco- exérèse). Le relevé du champ visuel révèle un important déficit périmétrique jouxtant le point de fixation (fig. B, partie supérieure du relevé). Cependant, la PIO est à 14 mmHg pour une épaisseur cornéenne centrale à 545 μm. La chambre antérieure est exagérément profonde et, en gonioscopie, le trabéculum est très pigmenté dans sa portion supérieure (fig. C) avec une hyperconcavité irienne. Il n’y a pas de fuseau de Krukenberg et l’iris n’est pas transilluminable, mais des dépôts pigmentés denses sont repérés au niveau de la zonule et de la hyaloïde antérieure (ligne de Scheie) à l’éclairement du fond d’œil (fig. D). Le diagnostic de glaucome pigmentaire éteint est donc évoqué. La cataracte est opérée et la PIO se maintient autour de 12 ± 2 mmHg durant huit années de suivi. Il est décidé un contrôle semestriel, sans traitement hypotenseur oculaire. La surveillance du champ visuel confirme la parfaite stabilité de l’atteinte périmétrique durant cette période, comme le montre l’évolution du VFI (fig. B). Le diagnostic de glaucome pigmentaire éteint est a priori (et a posteriori !) confirmé.

Fig. A Papille gauche.

Fig. B Champs visuels de 2001 à 2009 (programme GPA2 de Humphrey).

Fig. C Gonioscopie, portion supérieure de l’angle.

Fig. D Gonioscopie, ligne de Scheie.
L’administration de glucocorticoïdes exogènes (péri- ou intra-oculaires, topiques, oraux ou inhalés) ou certaines conditions d’hyperproduction endogène de glucocorticoïdes (syndrome de Cushing) sont susceptibles de causer une HTO cortico-induite, voire un glaucome secondaire à angle ouvert cortisonique (voir chapitre 14-IV). Cet état définit le terme de cortico-sensibilité [4, 5]. En cas d’administration topique oculaire d’une durée minimale de quatre à six semaines, une élévation de la PIO est observée chez 40 % de la population générale et chez 90 % des patients glaucomateux [5]. Cette cortico-sensibilité est un facteur de risque de développer un glaucome primitif à angle ouvert (GPAO) [23, 24].
Le glaucome cortisonique est très similaire au GPAO dans ses aspects cliniques et évolutifs.
Le mécanisme suspecté serait celui d’une augmentation de la résistance à l’écoulement de l’humeur aqueuse [2, 6, 7, 8, 30], en rapport avec des modifications morphologiques et fonctionnelles du réseau trabéculaire assez similaires à celles observées dans le GPAO [13].
L’hypertonie cortisonique est habituellement réversible à l’arrêt de l’administration des glucocorticoïdes [2, 6, 7], mais elle peut laisser des séquelles glaucomateuses proportionnelles à la durée de l’HTO, souvent bilatérales et symétriques. La possibilité de la constitution d’une HTO définitive est toutefois signalée lorsque la corticothérapie est prolongée plusieurs mois [15, 37], mais il est possible que ces cas représentent d’authentiques GPAO simplement précipités ou révélés par l’utilisation des corticoïdes.
A posteriori, seul un interrogatoire minutieux, voire la lecture d’anciennes ordonnances, permettront de rattacher un glaucome éteint à l’administration de glucocorticoïdes.
Le glaucome est l’une des complications les plus redoutables de l’uvéite. Environ 20 % des patients atteints d’uvéite aux États-Unis développent un glaucome. L’uvéite antérieure semble être plus pourvoyeuse d’HTO que les formes postérieures [9], bien que d’autres auteurs ne mettent pas en évidence de différence significative entre ces deux groupes [24].
Les mécanismes physiopathologiques de l’HTO au cours de l’uvéite sont encore mal connus. Il est admis que cette dernière modifie la dynamique de l’humeur aqueuse.
Ainsi, la PIO peut être augmentée par une majoration de la production d’humeur aqueuse, par une élévation de la résistance trabéculaire à l’évacuation de l’humeur aqueuse liée diversement à un œdème trabéculaire, à une constriction des cellules endothéliales trabéculaires sous l’effet de médiateurs inflammatoires tels que les rho-kinases [31], à un blocage trabéculaire direct (fibrine, protéines, débris ou cellules inflammatoires secondaires à la rupture de la barrière hémato-aqueuse), ou encore à un blocage du canal de Schlemm par des cellules inflammatoires.
La présence de prostaglandines intra-oculaires pourrait également être la cause d’une élévation de la PIO.
Enfin, de façon biomicroscopiquement évidente, un glaucome aigu par fermeture de l’angle peut être provoqué par un bloc pupillaire secondaire à la constitution de synéchies irido-cristalliniennes (« iris tomate »).
À l’inverse, la PIO peut diminuer par l’inflammation du corps ciliaire à l’origine d’une réduction de la production d’humeur aqueuse, ou par la choroïdite qui entraîne une augmentation de sa résorption uvéosclérale.
L’intrication de ces mécanismes peut être à l’origine de variations de la PIO, d’autant plus qu’il est difficile de faire la part entre une éventuelle HTO cortico-induite et celle liée à l’inflammation proprement dite [26, 35].
Les glaucomes uvéitiques éteints peuvent être uni- ou bilatéraux selon l’étiologie. Outre l’interrogatoire recherchant des antécédents d’uvéite, un examen attentif du patient s’attachera à retrouver des séquelles oculaires de poussées antérieures (synéchies antérieures ou postérieures, atrophie irienne, dépôts pigmentés sur la cristalloïde antérieure, foyers choriorétiniens, séquelles de vascularite, etc.).
Ils peuvent être la conséquence d’un traumatisme contusif ou pénétrant avec effraction de la paroi oculaire avec ou sans corps étranger intra-oculaire [3, 36]. Plusieurs mécanismes, parfois intriqués, sont proposés pour expliquer la survenue d’une élévation transitoire de la PIO :
l’obstruction trabéculaire par des hématies fraîches, dégénérées (glaucome à cellules fantômes ou ghost-cell glaucoma) ou phagocytées (glaucome hémolytique), des cellules inflammatoires, de la fibrine ou des débris ;
une blessure directe du trabéculum par élongation ;
un œdème trabéculaire inflammatoire.
Toutefois, cette poussée pressionnelle peut être iatrogène. Les procédures chirurgicales telles que la phaco-exérèse, la chirurgie vitréorétinienne ou la greffe de cornée sont parfois à l’origine d’une élévation transitoire de la PIO. Celle-ci est là encore provoquée par une augmentation de la résistance trabéculaire à l’évacuation de l’humeur aqueuse, résultant d’une libération de pigments, de la présence de cellules inflammatoires, d’un œdème trabéculaire inflammatoire et/ou de l’obstruction trabéculaire par des agents viscoélastiques dispersifs de haut poids moléculaire (hyaluronate de sodium), qui est moindre avec les agents viscoélastiques cohésifs (chondroïtine sulfate).
Mais, comme pour les uvéites, la responsabilité de l’utilisation postopératoire de corticoïdes locaux, parfois prolongée, doit toujours être évoquée.
La pression veineuse épisclérale (PVE) est un paramètre important de la régulation de la PIO. Sa valeur normale est de 8 à 10 mmHg. Une élévation de la PVE (> 10 mmHg) peut provoquer un collapsus du canal de Schlemm et une augmentation des résistances à l’écoulement de l’humeur aqueuse [18].
Un système complexe de vaisseaux lie le canal de Schlemm aux veines épisclérales qui drainent le sang jusqu’aux veines ciliaires antérieures et ophtalmique supérieure pour rejoindre enfin le sinus caverneux. Un obstacle mécanique au retour veineux oculaire peut être à l’origine d’une élévation de la PVE. Les causes les plus fréquentes d’obstruction veineuse sont :
l’orbitopathie dysthyroïdienne, manifestation extrathyroïdienne de la dysthyroïdite auto-immune (Basedow, Hashimoto). L’augmentation du volume des muscles et du compartiment graisseux par un infiltrat inflammatoire est à l’origine d’une stase veineuse orbitaire responsable d’une élévation de la PVE, mais aussi d’une élévation de la PIO par compression directe du globe oculaire. Le traitement étiologique ou une chirurgie de décompression orbitaire permettent de normaliser la PIO ;
les néoformations rétrobulbaires. Les tumeurs du système nerveux et des méninges, les tumeurs et masses vasculaires ou d’origine ORL, les masses inflammatoires, infectieuses et parasitaires, les tumeurs de la glande lacrymale, les tumeurs et kystes d’origine congénitale, les tumeurs primitives des parois orbitaires, les tumeurs mésenchymateuses primitives, les mélanomes, lymphomes, carcinomes ou métastases peuvent être à l’origine d’une stase veineuse et/ou comprimer de façon directe le globe oculaire dans un volume orbitaire inextensible, entraînant une élévation pathologique de la PIO. La réduction partielle ou totale du volume tumoral permet de restituer une PIO normale.
Elles sont à l’origine d’une élévation de la PVE par communication directe entre un système artériel à haut débit et un système veineux de bas débit. Les étiologies les plus fréquentes de malformation artérioveineuse sont les fistules artérioveineuses durales ou du sinus carotidocaverneux, les varices orbitaires et les hémangiomes faciaux (syndrome de Sturge-Weber).
Le traitement de la cause permet de restaurer une PVE normale et normaliser ainsi la PIO.
Le syndrome cave supérieur résulte d’une compression de la veine cave supérieure, compliquée ou non d’une thrombose. La compression est généralement d’origine tumorale maligne (néoplasmes bronchiques primitifs et lymphomes du médiastin) ou plus rarement bénigne (goitre plongeant). Le traitement définitif tenant compte de l’étiologie permet de lever l’obstacle au retour veineux et de normaliser ainsi la PVE.
Elle se manifeste par une apposition de l’iris périphérique au trabéculum. Les causes en sont multiples, entre autres :
iriennes : iris-plateau, kystes iriens ;
cristalliniennes : fermeture primitive de l’angle dans des globes biométriquement prédisposés par bloc pupillaire, luxation du cristallin ;
ciliaires : kystes ciliaires, œdème inflammatoire du corps ciliaire ;
choroïdiennes : turgescence, effusion, décollement ou tumeur choroïdienne (mélanomes uvéaux, carcinomes métastatiques) ;
sclérales : iatrogénique par indentation sclérale.
Elle entraîne une élévation de la PIO par diminution du drainage aqueux au travers de l’angle iridocornéen.
La soustraction de l’élément responsable restaure une ouverture optimale de l’angle iridocornéen et un drainage physiologique de l’humeur aqueuse, laissant parfois une neuropathie optique glaucomateuse séquellaire, dont la gravité est proportionnelle au degré et à la durée de la fermeture de l’angle.
Selon une hypothèse discutée, une hypersécrétion d’humeur aqueuse serait la conséquence d’une inflammation intra-oculaire par altération de la barrière hémato-aqueuse et par augmentation de la concentration protéique de l’humeur aqueuse créant un appel d’eau passif par effet osmotique.
Les crises glaucomatocyclitiques (syndrome de Posner-Schlossman), qui ont tendance à disparaître après 50 ans, sont accompagnées de poussées d’HTO sévères régressives dont la récurrence est à l’origine d’une neuropathie optique glaucomateuse dans 25 % des cas [1].
Ce mécanisme reste controversé. Plusieurs études suggèrent la possibilité d’une atrophie progressive du corps ciliaire secondaire à l’HTO chronique, entraînant une normalisation de la PIO [1, 19]. Les patients atteints de ce type de glaucome éteint seraient plus âgés et présenteraient des déficits anatomiques et campimétriques plus marqués [19].
La normalisation de la PIO s’expliquerait par la diminution concomitante de la production et de l’élimination de l’humeur aqueuse. La détermination du coefficient d’écoulement de l’humeur aqueuse appuierait ce diagnostic, objectivant une nette diminution de ce coefficient (< 0,1 μL/min/mmHg pour une valeur normale de 0,22 à 0,3 ml/min/mmHg) [19].
Le diagnostic différentiel entre glaucome éteint et glaucome à pression normale peut être très délicat en raison de l’existence de formes évolutives lentes. Seul un suivi prolongé structural et fonctionnel permet de repérer une évolution de la pathologie.
Les arguments suivants permettent habituellement de les distinguer d’une neuropathie optique glaucomateuse :
baisse d’acuité visuelle associée ou non à un déficit périphérique du champ visuel ;
altération du réflexe photomoteur (déficit pupillaire afférent, signe de Marcus-Gunn) ;
modification inconstante de la papille ou pâleur plus large que l’excavation ;
déficits périmétriques de systématisation différente, souvent sans respect du méridien horizontal ;
dyschromatopsie d’axe rouge-vert :
altération des potentiels évoqués visuels ;
antécédents personnels et familiaux d’atteinte neuro-ophtalmologique ou neurologique.
Les étiologies les plus fréquentes sont largement décrites dans les chapitres 13-I et 14-VII. Rappelons qu’elles peuvent être inflammatoires, infectieuses, vasculaires (neuropathie optique ischémique antérieure aiguë, choc hypovolémique), héréditaires (maladie de Leber), toxiques (alcooliques, médicamenteuses, etc.), compressives ou infiltratives, carentielles ou d’origine rétinienne (décollement, occlusion vasculaire, choriorétinite, etc.).
L’observateur ne devra pas se laisser abuser par de grandes excavations physiologiques et, au moindre doute, compléter le bilan par un examen périmétrique et éventuellement une évaluation précise de la papille et des fibres optiques par les analyseurs.
Le diagnostic différentiel avec un glaucome est plus difficile à poser lorsque la papille est en franche dysversion, d’autant que s’y associent des déficits périmétriques constitutionnels très similaires aux scotomes glaucomateux, et que les résultats des analyseurs y sont également trompeurs.
Les colobomes papillaires et les drusen du nerf optique, à l’origine de déficits périmétriques, sont plus facilement identifiables, en s’aidant éventuellement des techniques modernes d’imagerie.
La myopie, particulièrement lorsqu’elle est forte, engendre souvent des conflits diagnostiques qui peuvent ne jamais être complètement résolus, même avec les moyens d’évaluation les plus sophistiqués. La découverte de reliquats de dispersion pigmentaire chez des sujets myopes âgés complique encore l’interprétation étiologique des déficits périmétriques et l’évaluation de leur possible progression.
Les particularités papillaires mentionnées ci-dessus, le relevé de déficits périmétriques en réalité inexistants à cause de la mauvaise réalisation du relevé du champ visuel, la constatation d’une pachymétrie fine, des anomalies non spécifiques constatées sur les résultats des analyseurs… surtout s’ils sont plus ou moins associés, risquent de faire porter à tort un diagnostic de glaucome, qui s’avérerait naturellement non évolutif !
Le glaucome éteint est un diagnostic d’élimination, qui ne peut être affirmé que sur le long terme après un suivi identique à celui des GPAO. Il sous-entend que le diagnostic de glaucome ait initialement été établi de façon certaine. L’absence d’HTO, pondérée par la mesure de l’épaisseur cornéenne, peut être confirmée par la réalisation une courbe de PIO.
Le bilan étiologique repose sur la réalisation d’un interrogatoire très complet assorti d’un examen biomicroscopique minutieux.
L’absence de progression est établie en réalisant chaque fois une évaluation de l’état papillaire et des fibres nerveuses rétiniennes (rétinophotographies, analyseurs) et un relevé du champ visuel. En cas de découverte fortuite, il est recommandé de réaliser trois à quatre relevés périmétriques la première année afin de s’assurer de l’absence d’évolutivité de la maladie. Par la suite, le patient sera contrôlé de façon semestrielle, puis annuelle, à la recherche chaque fois d’une possible progression.
En théorie, puisque le glaucome est éteint, aucun traitement n’est indiqué. Toutefois, lors des premières consultations et avec l’absence du recul, il est quasiment impossible d’être affirmatif sur la non-évolutivité du glaucome diagnostiqué. Un traitement hypotonisant oculaire sera donc institué par principe de précaution… quitte à le supprimer ultérieurement, lorsque l’anamnèse et les résultats des examens successifs auront convaincu le médecin qu’il se trouve en présence d’un glaucome réellement éteint. Cependant, en tout état de cause, même si cette certitude paraît acquise, le patient doit continuer à être régulièrement examiné, au moins annuellement.
Retenir
Le glaucome éteint est une neuropathie optique glaucomateuse avec des déficits périmétriques caractéristiques et une PIO normale, non évolutive.
Les étiologies les plus fréquentes sont le glaucome pigmentaire, cortisonique, uvéitique ou traumatique.
Parmi les diagnostics différentiels on retient le GPN, les neuropathies optiques non glaucomateuses et les erreurs de diagnostic.
Traitement : aucun n’est nécessaire en théorie, mais en pratique institué au moment du diagnostic quitte à le supprimer ultérieurement.
Le suivi est annuel avec PIO, fond d’œil, champ visuel et, éventuellement, analyse de la structure s’il s’agit d’un glaucome débutant.
[1] Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011 ; 59 : S97-101.
[2] Armaly MF. Effect of corticosteroids on intraocular pressure and fluid dynamics. II. The effect of dexamethasone in the glaucomatous eye. Arch Ophthalmol. 1963 ; 70 : 492-9.
[3] Bai HQ, Yao L, Wang DB, et al. Causes and treatments of traumatic secondary glaucoma. Eur J Ophthalmol. 2009 ; 19 : 201-6.
[4] Becker B. Intraocular pressure response to topical corticosteroids. Invest Ophthalmol. 1965 ; 4 : 198-205.
[5] Becker B, Ballin N. Glaucoma and corticosteroid provocative testing. Arch Ophthalmol. 1965 ; 74 : 621-4.
[6] Becker B, Mills DW. Corticosteroids and intraocular pressure. Arch Ophthalmol. 1963 ; 70 : 500-7.
[7] Becker B, Mills DW. Elevated intraocular pressure following corticosteroid eye drops. JAMA. 1963 ; 185 : 884-6.
[8] Bernstein HN, Schwartz B. Effects of long-term systemic steroids on ocular pressure and tonographic values. Arch Ophthalmol. 1962 ; 68 : 742-53.
[9] Bodaghi B, Cassoux N, Wechsler B, et al. Chronic severe uveitis : etiology and visual outcome in 927 patients from a single center. Medicine. 2001 ; 80 : 263-70.
[10] Buller C, Johnson DH, Tschumper RC. Human trabecular meshwork phagocytosis. Observations in an organ culture system. Invest Ophthalmol Vis Sci. 1990 ; 31 : 2156-63.
[11] Campbell DG. Pigmentary dispersion and glaucoma. A new theory. Arch Ophthalmol. 1979 ; 97 : 1667-72.
[12] Campbell DG, Jeffrey CP. Pigmentary dispersion in the human eye, SEM. Scan Electron Microsc. 1979 ; 3 : 329-33.
[13] Clark AF, Wordinger JR. The role of steroids in outflow resistance. Exp Eye Res. 2009 ; 88 : 752-9.
[14] Collaborative Normal-Tension Glaucoma Study Group. CNTGS. Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. Am J Ophthalmol. 1998 ; 126 : 487-97.
[15] Espildora J, Vicuna P, Diaz E. Cortisone-induced glaucoma : a report on 44 affected eyes. J Fr Ophtalmol. 1981 ; 4 : 503-8.
[16] Fuchshofer R, Tamm ER. Modulation of extracellular matrix turnover in the trabecular meshwork. Exp Eye Res. 2009 ; 88 : 683-8.
[17] Gottanka J, Johnson DH, Grehn F, Lutjen-Drecoll E. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. J Glaucoma. 2006 ; 15 : 142-51.
[18] Greenfield DS. Glaucoma associated with elevated episcleral venous pressure. J Glaucoma. 2000 ; 9 : 190-4.
[19] Greve EL. Glaucoma symposium, diagnosis and therapy : Amsterdam, September 1979 : with panel discussions on glaucoma and cataract, narrow angles. Documenta Ophthalmologica Proceedings, 1980, series 22.
[20] Johnson M. What controls aqueous humour outflow resistance ? Exp Eye Res. 2006 ; 82 : 545-57.
[21] Karickhoff JR. Pigmentary dispersion syndrome and pigmentary glaucoma : a new mechanism concept, a new treatment, and a new technique. Ophthalmic Surg. 1992 ; 23 : 269-77.
[22] Keller KE, Aga M, Bradley JM, et al. Extracellular matrix turnover and outflow resistance. Exp Eye Res. 2009 ; 88 : 676-82.
[23] Kitazawa Y, Horie T. The prognosis of corticosteroid-responsive individuals. Arch Ophthalmol. 1981 ; 99 : 819-23.
[24] Lewis JM, Priddy T, Judd J, et al. Intraocular pressure response to topical dexamethasone as a predictor for the development of primary open-angle glaucoma. Am J Ophthalmol. 1988 ; 106 : 607-12.
[25] Lichter PR, Shaffer RN. Diagnostic and prognostic signs in pigmentary glaucoma. Trans Am Acad Ophthalmol Otolaryngol. 1970 ; 74 : 984-98.
[26] Moorthy RS, Mermoud A, Baerveldt G, et al. Glaucoma associated with uveitis. Surv Ophthalmol. 1997 ; 41 : 361-94.
[27] Murphy CG, Johnson M, Alvarado JA. Juxtacanalicular tissue in pigmentary and primary open angle glaucoma. The hydrodynamic role of pigment and other constituents. Arch Ophthalmol. 1992 ; 110 : 1779-85.
[28] Neri P, Azuara-Blanco A, Forrester JV. Incidence of glaucoma in patients with uveitis. J Glaucoma. 2004 ; 13 : 461-5.
[29] American Academy of Ophthalmology. Glaucome. 2010.
[30] Paterson G. Studies of the response to topical dexamethasone of glaucoma relatives. Trans Ophthalmol Soc UK. 1965 ; 85 : 295-305.
[31] Rao PV, Deng PF, Kumar J, Epstein DL. Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y27632. Invest Ophthalmol Vis Sci. 2001 ; 42 : 1029-37.
[32] Ritch R. Nonprogressive lowtension glaucoma with pigmentary dispersion. Am J Ophthalmol. 1982 ; 94 : 190-6.
[33] Ritch R. A unification hypothesis of pigment dispersion syndrome. Trans Am Ophthalmol Soc. 1996 ; 94 : 381-409.
[34] Scheie HG, Fleischhauer HW. Idiopathic atrophy of the epithelial layers of the iris and ciliary body ; a clinical study. AMA Arch Ophthalmol. 1958 ; 59 : 216-28.
[35] Siddique SS, Suelves AM, Baheti U, Foster CS. Glaucoma and uveitis. Surv Ophthalmol. 2013 ; 58 : 1-10.
[36] Sihota R, Sood NN, Agarwal HC. Traumatic glaucoma. Acta Ophthalmol Scand. 1995 ; 73 : 252-4.
[37] Spaeth GL, Rodrigues MM, Weinreb S. Steroidinduced glaucoma : A. Persistent elevation of intraocular pressure B. Histopathological aspects. Trans Am Ophthalmol Soc. 1977 ; 75 : 353-81.
[38] Sugar HS, Barbour FA. Pigmentary glaucoma ; a rare clinical entity. Am J Ophthalmol. 1949 ; 32 : 90-2.
[39] Sugiura T, Ito M, Mizokami K. A comparative study of optic disc appearances in progressive and non-progressive lowtension glaucoma. Nihon Ganka Gakkai Zasshi. 1991 ; 95 : 343-7.
[40] Tamm ER, Fuchshofer R. What increases outflow resistance in primary openangle glaucoma ? Surv Ophthalmol. 2007 ; 52 : S101-4.
[41] Tektas OY, LutjenDrecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res. 2009 ; 88 : 769-75.
E. Blumen-Ohana
Les glaucomes secondaires à angle ouvert sont des entités particulières qu’il faut savoir évoquer devant toute neuropathie optique glaucomateuse : la prise en charge du primum movens peut être cruciale dans certains cas.
Les uvéites sont susceptibles de se compliquer d’hypertension oculaire (HTO) et de glaucome, imposant souvent un traitement spécifique et une vigilance vis-à-vis des traitements anti inflammatoires stéroïdiens.
Le glaucome cortico-induit est une entité connue à redouter.
La notion de traumatisme doit être recherchée devant tout glaucome unilatéral. La gonioscopie peut permettre d’identifier une sémiologie évocatrice.
Une chirurgie de cataracte compliquée ou congénitale peut être à l’origine d’un glaucome à angle ouvert. La vigilance est donc de mise lors du suivi de ces patients en dépit de la date opératoire initiale.
Les glaucomes secondaires à angle ouvert recouvrent différentes entités qui aboutissent à un dysfonctionnement trabéculaire avec son cortège d’HTO et de neuropathie optique progressive. Les glaucomes pigmentaire et exfoliatif sont traités par ailleurs. Ce chapitre concerne les autres glaucomes secondaires fréquents : le glaucome uvéitique, le glaucome cortico-induit, le glaucome post-traumatique et le glaucome de l’aphake.
Les uvéites, pathologies inflammatoires intra-oculaires, représentent une cause de handicap visuel non négligeable. Elles sont à l’origine d’environ 10 % des cas de cécité légale dans le monde, représentant ainsi la 5e cause de cécité mondiale [74, 85]. Le terme d’uvéite se rapporte à des entités cliniques variées avec des étiologies différentes. Le terme de glaucome uvéitique fait référence à la neuropathie optique progressive secondaire à une uvéite entraînant elle-même une HTO. Prendre en charge un glaucome uvéitique impose d’identifier l’origine du processus inflammatoire ainsi que le mécanisme de l’HTO, et de traiter de concert l’inflammation et le facteur pressionnel intra-oculaires.
La prévalence du glaucome uvéitique a été diversement appréciée dans les études disponibles, ce chiffre pouvant varier du simple au double en fonction des critères diagnostiques choisis, de l’origine et du siège de l’uvéite, de sa durée d’évolution, de la nécessité d’un traitement corticoïde plus ou moins long ou répété, etc. On retrouve ainsi un glaucome dans 8 à 20 % des uvéites [22, 41, 56, 65], dont l’incidence augmente avec la durée d’évolution de l’uvéite [42, 63] ou la prise d’une corticothérapie, une étude rapportant la survenue d’un glaucome dans 45 % des cas en cas d’implant intravitréen délivrant un corticoïde [11]. Certaines étiologies sont de plus grandes pourvoyeuses d’HTO : une étude rapporte la survenue de 37 % de cas d’HTO au cours d’uvéites herpétiques [69]. L’âge, l’origine ethnique ou le sexe ne sont pas des facteurs favorisant la survenue d’un glaucome uvéitique [62].
Les mécanismes physiopathologiques induisant une HTO et un glaucome uvéitique ne sont pas tous univoques. Initialement, à la phase inflammatoire aiguë, on observe souvent une hypotonie oculaire résultant d’une hyposécrétion, induite par l’inflammation du corps ciliaire, et d’une augmentation du flux uvéoscléral. Un déséquilibre entre la production et l’évacuation de l’humeur aqueuse est vraisemblable, s’installant progressivement et conduisant à une HTO puis à un glaucome secondaire. Le diagnostic peut poser problème avec les interférences thérapeutiques, notamment avec les HTO cortico-induites.
Un glaucome uvéitique peut être à angle iridocornéen ouvert ou fermé. La forme la plus fréquente est le glaucome uvéitique à angle ouvert, où l’on observe une résistance à l’écoulement de l’humeur aqueuse « mécanique » par encombrement du trabéculum, (pigments, fibrine, protéines et cellules inflammatoires). Une trabéculite, ou plus généralement un dysfonctionnement trabéculaire induit par le processus inflammatoire, peut être aussi en cause [62]. D’autres facteurs ont pu être impliqués dans la survenue de l’HTO secondaire à une inflammation intra-oculaire : c’est le cas des rho-kinases et des prostaglandines [72] avec, à moyen terme, des phénomènes chroniques s’installant au niveau du trabéculum et entravant son fonctionnement [86]. Les cytokines impliquées dans l’inflammation peuvent également favoriser la prolifération de membranes fibrovasculaires au niveau de l’angle iridocornéen. La prise en charge des phénomènes inflammatoires implique régulièrement une corticothérapie, à l’origine elle-même d’une HTO dans 30 % des cas [8], par le biais d’une diminution de l’évacuation de l’humeur aqueuse. La survenue d’une HTO dans ce contexte est soumise à une susceptibilité individuelle [33], au type de corticoïde utilisé, à son mode d’administration ainsi qu’à la durée du traitement.
Dans le cadre des uvéites, on peut également observer le développement de glaucomes par fermeture de l’angle : synéchies iridocristalliniennes postérieures risquant d’aboutir à un bloc pupillaire post-inflammatoire avec parfois un « iris tomate » ; synéchies antérieures périphériques ; effusion inflammatoire avec bascule antérieure du corps ciliaire pouvant réaliser l’équivalent d’un iris plateau [52].
Les mécanismes physiopathologiques pouvant conduire à une HTO dans le cadre d’une uvéite sont résumés en figure 14-12.
Fig. 14-12 Facteurs susceptibles d’influencer la PIO dans les uvéites.
Certaines uvéites sont particulièrement susceptibles d’entraîner une HTO et éventuellement un glaucome uvéitique : uvéites herpétiques, uvéites survenant dans le cadre d’arthrite chronique juvénile [34], syndrome de Posner-Schlossman, iridocyclite hétérochromique de Fuchs [56].
Elles sont plus volontiers induites par l’Herpes simplex virus de type 1. On peut dans ce contexte observer une atteinte cornéenne évocatrice, qu’elle soit épithéliale, disciforme, stromale ou endothéliale, associée à l’uvéite. Une uvéite peut être également le fruit d’une réactivation du virus varicelle-zoster par le biais d’un zona ophtalmique. Les lésions cutanées en secteur sont évocatrices, et l’atteinte oculaire est suspectée quand la branche ophtalmique du trijumeau est concernée. Dans ce contexte, la présence de précipités rétrodescemétiques ou de zones d’atrophie irienne est évocatrice. La prise en charge impose la mise en route d’un traitement antiviral effectif, avant tout traitement anti-inflammatoire ; la pression intra-oculaire (PIO) peut alors évoluer favorablement ou nécessiter une prise en charge propre. Les formes récidivantes ou chroniques imposent des mesures préventives, souvent un traitement antiviral poursuivi longtemps, voire en continu [44, 53]. Ce traitement antiviral est régulièrement associé à un traitement corticoïde pour limiter l’inflammation ; une surveillance étroite et régulière s’impose dans ce contexte avec monitoring de la PIO, et un traitement hypotonisant oculaire médical, voire chirurgical, est souvent nécessaire.
L’uvéite est retrouvée jusque dans 20 % des cas d’arthrite chronique juvénile, en règle bilatérale et classiquement non granulomateuse [40, 70].
Il est aussi connu sous le nom de crise glaucomatocyclitique et caractérisé par des crises d’HTO souvent spectaculaires contrastant avec une inflammation intra-oculaire modérée. Les signes sont généralement unilatéraux : sensation de halos et gêne visuelle associées à une HTO, quelques précipités rétrodescemétiques fins blanchâtres, Tyndall de chambre antérieure peu marqué. Il existe parfois un œdème cornéen, induit par la crise d’HTO. La prise en charge repose sur la gestion des crises d’HTO avec parfois l’adjonction prudente d’anti-inflammatoires en se méfiant de la survenue d’un glaucome cortico-induit [78].
Il s’agit d’une uvéite chronique, unilatérale en règle, associée à une hétérochromie irienne touchant l’adulte jeune, sans prédominance de sexe. Une cataracte survient souvent en cours d’évolution, et l’HTO et le glaucome sont également des complications classiquement décrites. L’atteinte vitréenne est exceptionnelle. La prévalence du glaucome dans l’iridocyclite de Fuchs peut atteindre près de 60 % des cas [46, 47]. Certains signes sont très évocateurs, notamment l’hétérochromie, les précipités rétrodescemétiques « en étoile », la cataracte associée, etc. (fig. 14-13). À noter une réponse thérapeutique fréquente au traitement par anti-inflammatoires non stéroïdiens, alors que les corticoïdes sont habituellement peu efficaces.

Fig. 14-13 Iridocyclite hétérochromique de Fuchs.
D’autres étiologies d’uvéite sont susceptibles d’être associées à une HTO et à un glaucome secondaire (fig. 14-14) : choriorétinite toxoplasmique, syphilis, sarcoïdose et maladie de Behçet.
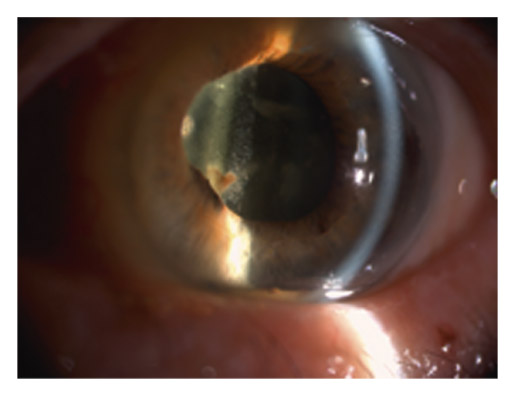
Fig. 14-14 Glaucome uvéitique. (Cliché : J.-P. Renard.)
L’HTO survenant dans un contexte d’uvéite peut être en rapport avec le processus inflammatoire, un effet secondaire de la corticothérapie ou encore provoqué par une fermeture de l’angle iridocornéen.
Elle repose avant tout sur le traitement étiologique de la cause éventuelle. Cet aspect de la prise en charge est important en cas d’étiologie infectieuse et notamment virale suspectée. Dans l’iridocyclite de Fuchs, certains virus ont été incriminés, particulièrement celui de la rubéole, expliquant qu’on ne propose une corticothérapie qu’en cas de baisse d’acuité visuelle significative. Le syndrome de Posner-Schlossman est une inflammation intra-oculaire récidivante dont la cause n’est pas clairement identifiée ; l’étiologie virale ne pouvant être complètement écartée, on peut être amené à proposer un traitement antiviral dans certaines formes cliniques.
La gestion de l’inflammation passe régulièrement par une corticothérapie locale et/ou systémique. Certaines formes galéniques permettent d’éviter les conservateurs, ce qui peut être intéressant en cas de traitement prolongé. L’utilisation d’anti-inflammatoires non stéroïdiens peut être justifiée dans certains cas, de même que certains immunosuppresseurs qui autorisent alors une épargne cortisonique. Une dilatation pupillaire peut être recommandée et sera en particulier pratiquée lors de chaque contrôle, afin d’éviter la constitution de synéchies postérieures, facteurs aggravant l’HTO ou le glaucome préexistant.
Le traitement médical va privilégier l’utilisation de bêtabloquants et d’inhibiteurs de l’anhydrase carbonique qui limitent la production d’humeur aqueuse. Les α2-agonistes peuvent également être proposés dans ce contexte mais sont souvent pourvoyeurs d’effets secondaires. Les analogues de prostaglandines seront prescrits avec précaution car susceptibles d’interférer avec le processus inflammatoire, voire d’entraîner une récidive d’herpès oculaire ou un œdème maculaire [80, 90].
Les alternatives chirurgicales varient en fonction de la sévérité du tableau clinique, allant de la réalisation d’une iridotomie périphérique au laser YAG pour limiter la constitution de synéchies antérieures périphériques en cas de blocage pupillaire, à la mise en place de dispositifs de drainage ou au cyclo-affaiblissement en cas de glaucome réfractaire. La trabéculoplastie sélective a été proposée par certaines équipes en cas d’angle iridocornéen ouvert, avec des résultats et des conséquences acceptables [80]. Les chirurgies filtrantes réalisées dans un contexte d’uvéite doivent être encadrées par un traitement anti-inflammatoire, afin d’en optimiser les résultats [12]. C’est ainsi que l’administration d’une corticothérapie par voie systémique en péri-opératoire peut être indiquée [23]. L’utilisation d’antimétabolites au cours d’une trabéculectomie ou d’une sclérectomie profonde non perforante [1, 31] permet d’augmenter le taux de succès chirurgical [15]. Le glaucome uvéitique pouvant être considéré à risque d’échec chirurgical, certains auteurs proposent la mise en place de dispositifs de drainage [16, 61] ; ils permettent un meilleur contrôle de la PIO, au prix parfois de complications non négligeables. Le cyclo-affaiblissement au laser diode n’est pas anodin sur un corps ciliaire déjà fragilisé par les phénomènes inflammatoires, et il sera réservé, en dernier recours, aux glaucomes réfractaires, avec parcimonie en raison du risque de phtyse [5].
L’HTO induite par une corticothérapie est une situation clinique décrite depuis les années 1950 [54], qui peut survenir quelle que soit la voie d’administration des corticoïdes – topique, intravitréenne [50], locorégionale ou systémique.
Certaines personnes sont à risque de développer une HTO, voire un glaucome induit par une corticothérapie :
L’HTO cortico-induite semble résulter d’une résistance à l’écoulement trabéculaire de l’humeur aqueuse. Les mécanismes de survenue de cette résistance ne sont pas univoques, mais on évoque une modification du filtre trabéculaire [21] par les corticoïdes sous certaines conditions, laissant envisager une prédisposition génétique. Les corticoïdes engendrent des modifications structurales au niveau trabéculaire par le biais de dépôts de matériel extracellulaire [92, 93] (glycosaminoglycanes, fibronectine, etc.) et par le biais d’une inhibition des propriétés phagocytaires et protéolytiques, en particulier des métalloprotéinases [82, 92]. La prédisposition génétique suspectée a permis d’identifier certains gènes susceptibles d’intervenir dans l’HTO cortico-induite, notamment le gène de la myociline (anciennement TIGR) [64, 92], même si les relations de cause à effet ne sont pas encore démontrées formellement [49]. Le degré d’HTO est corrélé à la puissance anti-inflammatoire du corticoïde considéré [14] : la prednisolone et la dexaméthasone induisent plus facilement une HTO que la fluorométholone ou la rimexolone.
L’HTO apparaît classiquement dans les semaines qui suivent le début d’un traitement topique [2, 3, 14], mais peut néanmoins survenir plus rapidement, dans les heures suivant la mise en route du traitement topique [91] ou systémique [35]. Elle est plus fréquente en cas d’administration topique du corticoïde, comparativement à une administration générale [35]. Il a été rapporté une élévation rapide de la PIO après injection intravitréenne dans près de 50 % des cas [45] ; certains auteurs préconisent la mise en place d’un protocole de surveillance stricte de la PIO débutant dans l’heure suivant l’administration intravitréenne du corticoïde [50].
Les signes cliniques du glaucome cortico-induit sont similaires à ceux du GPAO, avec la notion d’un antécédent de corticothérapie. Les anomalies papillaires et du champ visuel, constatées parfois a posteriori, sont directement liées à l’HTO cortico-induite.
Pendant toute la durée de la corticothérapie, la surveillance passe par un contrôle régulier de la PIO associé à un dépistage de la neuropathie optique glaucomateuse [49].
En cas d’HTO constatée, il faut chercher à interrompre la corticothérapie [91]. S’il n’est pas possible de l’interrompre complètement, on pourra envisager la prise d’une molécule moins hypertonisante oculaire, proposer des anti-inflammatoires non stéroïdiens ou des immunosuppresseurs afin de diminuer la charge de corticoïdes administrée. Suspendre une corticothérapie peut signifier devoir réaliser l’évacuation chirurgicale de cristaux de corticoïdes retard en intravitréen [43].
En cas de nécessité, l’utilisation des traitements hypotonisants oculaires se fera sur le même schéma que dans le GPAO avec une escalade thérapeutique pouvant aller jusqu’à la réalisation d’une chirurgie filtrante [38, 45] ; certains auteurs ont également proposé une trabéculoplastie au laser, avec un résultat positif, mais sur un nombre restreint de patients [50, 75]. Des lésions irréversibles à l’arrêt de la corticothérapie sont observées dans moins de 5 % des cas d’HTO cortico-induite [32, 35], à condition de respecter ces règles rigoureuses de suivi clinique.
La prise en charge de toute corticothérapie repose sur la surveillance de la PIO, rapidement, dans les cinq à sept jours après l’initiation du traitement puis régulièrement tout au long de celui-ci.
Les traumatismes oculaires sont pourvoyeurs de complications diverses pouvant survenir au niveau de toutes les structures de l’œil. La notion même de traumatisme à l’interrogatoire doit alerter le clinicien et orienter son examen à la recherche de ces complications potentielles qui peuvent survenir des années après le traumatisme initial. Parmi celles-ci, le glaucome post-traumatique a un spectre particulièrement pernicieux, dans la mesure où, comme toute neuropathie optique glaucomateuse, il peut évoluer longtemps avant que n’apparaissent les premiers signes fonctionnels. Il est donc fondamental de rechercher la notion de traumatisme dans les antécédents personnels de tout patient.
La prévalence et l’incidence du glaucome post-traumatique ne sont pas des données facilement accessibles. Ils représenteraient 23 à 26 % des glaucomes secondaires, et 2 à 10 % de l’ensemble des glaucomes [48, 58]. Néanmoins les traumatismes oculaires sont relativement fréquents avec des caractéristiques épidémiologiques connues [13]. Le terrain privilégié de survenue est celui de l’homme jeune en pleine activité professionnelle.
Les forces impliquées lors d’une contusion oculaire vont induire une déformation du globe oculaire avec un rétrécissement antéro-postérieur et une expansion pré-équatoriale. Un traumatisme contusif peut être à l’origine d’atteinte des différentes structures intra-oculaires : les forces provenant du traumatisme initial vont être transférées dans plusieurs directions avec des répercussions à chaque niveau du globe oculaire [13, 73].
Les traumatismes contusifs induisent au stade initial, sous l’influence des forces mécaniques initiées, une augmentation de la pression en chambre antérieure, avec un déplacement postérieur du bloc iridocristallinien et des étirements, voire des déchirures, de structures vascularisées comme l’iris et le corps ciliaire [30, 73].
L’hyphéma est une complication fréquente des traumatismes oculaires et peut survenir jusque dans 60 % des cas [26, 28, 37]. Il correspond à l’apparition d’une collection de sang entre la cornée et l’iris, et peut être classé en quatre stades suivant son importance : stade I inférieur au tiers de la chambre antérieure, stade II compris entre le tiers et la moitié de la chambre antérieure, stade III dépassant la moitié de la chambre antérieure, le stade IV correspondant à un envahissement total de la chambre antérieure. L’hyphéma à lui seul peut engendrer une HTO, en particulier s’il est important [26, 30]. Il peut également être à l’origine de complications susceptibles de créer une HTO : c’est le cas des lésions trabéculaires, des synéchies iridocristalliniennes avec éventuel blocage pupillaire, des goniosynéchies, etc. Une récidive d’hyphéma jusque dans 38 % des cas, entre le 2e et le 5e jour après le traumatisme, a un pronostic plus péjoratif encore [89, 73] : il peut se compliquer à nouveau de synéchies ou d’HTO, mais ne semble pas altérer de façon significative la fonction visuelle à terme, par rapport aux yeux qui ont présenté un saignement unique. Un terrain particulier à rechercher, favorisant la survenue d’hyphéma, est celui des patients présentant une thalassémie, chez qui les hématies déformées sont difficilement évacuées au niveau du trabéculum (fig. 14-15).
Des phénomènes inflammatoires peuvent être déclenchés par la contusion oculaire, avec libération de médiateurs, notamment de prostaglandines, expliquant alors une phase d’hypotonie intra-oculaire initiale. Il n’est pas rare d’observer, au stade aigu, un effet de Tyndall en chambre antérieure, des précipités rétrodescemétiques, voire une trabéculite ; ces signes peuvent se chroniciser et entraîner des synéchies antérieures et postérieures, à l’origine d’une éventuelle fermeture progressive de l’angle iridocornéen, avec son cortège de complications propres et le développement d’un glaucome secondaire par fermeture de l’angle.
Une récession angulaire peut être observée au décours immédiat d’un épisode contusif ou après évacuation d’un hyphéma qui limitait l’analyse de l’angle iridocornéen. On visualise alors un élargissement irrégulier de la bande ciliaire. La PIO peut être normale dans les suites immédiates du traumatisme, voire durant de nombreuses années. Un glaucome lié à une récession de l’angle peut survenir parfois plusieurs décennies après le traumatisme initial. Le patient concerné devra être prévenu de cette possible évolution, si l’examen initial révèle une récession angulaire (fig. 14-16). En effet, il semble que cette situation puisse prédisposer à la survenue d’un GPAO, d’autant plus que l’étendue de la récession est importante [73, 76] ou qu’il existe une réponse positive aux corticoïdes [83], voire un GPAO sur l’œil adelphe [76, 87].
L’angle iridocornéen peut être modifié ; on peut observer une iridodialyse, une déchirure de la racine de l’iris ou du trabéculum, une cyclodialyse qui fait communiquer la chambre antérieure et l’espace suprachoroïdien, avec parfois une PIO temporairement abaissée au stade aigu.
Des modifications cristalliniennes peuvent apparaître dès le stade initial de la contusion oculaire, allant de la sub-luxation à la luxation totale avec retentissement cornéen ou vitréorétinien. Les modifications cristalliniennes sont susceptibles d’entraîner une HTO par le biais d’un blocage pupillaire, d’une issue de vitré réalisant un obstacle trabéculaire, voire des mécanismes phakolytiques ou phako-antigéniques (fig. 14-17).
Enfin, le tableau clinique peut se compléter d’atteintes concomitantes du segment postérieur, comme une hémorragie intravitréenne, une déhiscence rétinienne, voire un décollement de rétine.
Une hémorragie intra-oculaire importante peut se compliquer ultérieurement d’un glaucome hémolytique secondaire à une obstruction trabéculaire par des débris inflammatoires, des macrophages et des hématies. Un glaucome hémosidérotique peut survenir également au décours d’un épisode hémorragique (hémoglobine phagocytée par les cellules trabéculaires, avec altérations fonctionnelles). Citons enfin le glaucome à cellules fantômes, qui survient dans les suites d’une hémorragie intravitréenne, où les hématies vieillies se rigidifient et passent au travers de la hyaloïde antérieure et de la zonule, en chambre antérieure, où elles peuvent constituer un pseudo-hypopion, puis au travers du trabéculum qu’elles encrassent, constituant un obstacle mécanique. La survenue d’un glaucome à cellules fantômes peut survenir quelques semaines, voire des années, après le traumatisme initial.

Fig. 14-15 Dialyse irienne et hyphéma post-contusif. (Cliché : J.-P. Renard.)

Fig. 14-16 Récession angulaire post-contusive. (Cliché : J.-P. Renard.)
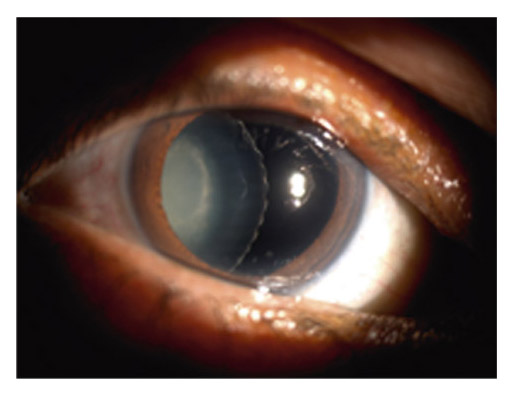
Fig. 14-17 Luxation post-traumatique du cristallin. (Cliché : J.-P. Renard.)
Outre les données de l’interrogatoire, l’examen clinique peut orienter vers un antécédent de contusion oculaire : irrégularité pupillaire, dialyse irienne, sub-luxation du cristallin, glaucome unilatéral, etc. Ces signes d’appel encourageront à réaliser une gonioscopie qui pourra retrouver des lésions angulaires évocatrices. Toutes les complications décrites pour le stade aigu sont susceptibles de se manifester à distance du traumatisme responsable. L’examen ophtalmologique devra être orienté, bilatéral et comparatif, avec examen gonioscopique et du fond d’œil avec dilatation. Les lésions angulaires les plus fréquemment retrouvées rétrospectivement sont la récession angulaire, les déchirures du trabéculum et les lésions de la racine de l’iris [76].
Les traumatismes perforants ont le même terrain de survenue, à savoir l’homme jeune en pleine activité professionnelle. Ils posent le problème de la gestion de l’urgence et de la reconstitution ad integrum des structures anatomiques. Les plaies peuvent être multiples, de grande taille, s’associer à des plaies du cristallin, ou des corps étrangers intra-oculaires nécessitant une prise en charge spécifique.
La pression intra-oculaire peut augmenter secondairement, en raison des lésions induites par le traumatisme perforant initial, qu’elles soient d’ordre inflammatoire, hémorragique ou anatomique, comme dans les traumatismes purement contusifs. La constitution de membranes cyclitiques, l’avancée du bloc iridocristallinien et la survenue de synéchies peuvent favoriser la survenue d’une hypertonie oculaire dans ce contexte. Une éventuelle invasion épithéliale peut également intervenir dans la survenue d’une hypertonie oculaire ; le risque d’ignorer un corps étranger intra-oculaire, éventuellement métallique, pourra entraîner une sidérose ou une chalcose.
La prise en charge chirurgicale au stade aigu doit être complète, restaurer toutes les structures anatomiques et la circulation de l’humeur aqueuse au niveau de la pupille et de l’angle iridocornéen [73]. On s’attachera ainsi à évacuer tout encombrement au niveau du segment antérieur, libérer les synéchies favorisées par l’hypothalamie prolongée, reconstruire et maintenir l’iris à distance de la cornée, libérer l’angle iridocornéen, etc. (fig. 14-18).
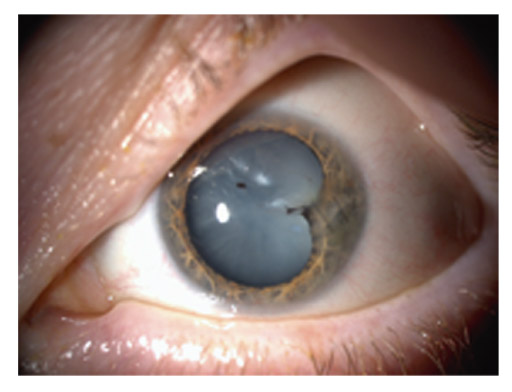
Fig. 14-18 Séquelles de plaie oculaire avec synéchies antérieures et postérieures. (Cliché : J.-P. Renard.)
Le glaucome secondaire à des brûlures chimiques survient après une exposition à un produit acide ou, plus fréquemment, à un produit basique [66]. Le spectre des complications induites par une projection de produit basique est important, avec un effet délétère prolongé du produit considéré. À la phase précoce, une toxicité directe du produit sur le trabéculum ou les voies d’évacuation de l’humeur aqueuse, la réaction inflammatoire ou l’augmentation du flux sanguin uvéal peuvent expliquer la survenue d’une HTO. Le lavage abondant et précoce s’impose pour tenter de limiter ces effets destructeurs. À noter également la possibilité d’une hypotonie majeure en cas de dommage direct du corps ciliaire. À la phase tardive, le tableau clinique associe une HTO à de nombreuses lésions, en particulier cornéennes. Les analogues de prostaglandines ainsi que les myotiques seront évités, les collyres sans conservateur sont à privilégier, les corticoïdes controversés, et la chirurgie filtrante souvent vouée à l’échec. En cas de prise en charge chirurgicale nécessaire, la mise en place de tubes de drainage ou la cyclophotocoagulation pourront être proposées [30, 66].
Des hémorragies sous-conjonctivales sont toujours susceptibles de cacher une plaie perforante du globe oculaire.
Un autre piège diagnostique dans ce contexte est représenté par une sub-luxation minime du cristallin éventuellement associée à une petite hernie vitréenne responsable d’une HTO secondaire évoluant à bas bruit.
Le glaucome post-traumatique est en soi un piège diagnostique à envisager devant tout glaucome unilatéral.
Il est capital d’assurer un bilan et une prise en charge optimale d’emblée de tout traumatisme oculaire : recherche de corps étranger intra-oculaire, parage et suture en étant le plus conservateur possible, couverture antibiotique, anti-inflammatoire et cycloplégique, traitement de l’hyphéma [27, 29].
Si la PIO ne peut pas être maîtrisée, la réalisation précoce d’une chirurgie filtrante s’impose [55, 57].
La prise en charge du glaucome post-traumatique est superposable à celle de toute neuropathie optique glaucomateuse, nécessitant :
une baisse de la PIO. Le traitement chirurgical est classiquement celui d’un glaucome présentant des facteurs de risque d’échec de la chirurgie filtrante et on y associera volontiers des anti-métabolites [55, 58]. Les autres options chirurgicales (tubes de drainage, cyclodestruction) seront discutées au cas par cas. La trabéculoplastie est en général inefficace au niveau d’un angle iridocornéen remanié par un traumatisme ;
un traitement spécifique d’un éventuel obstacle à l’évacuation de l’humeur aqueuse, à distance du traumatisme initial (iridotomie périphérique, iridoplastie, etc.) ;
une gestion des éventuelles lésions associées, notamment vitréorétiniennes.
La notion de contusion oculaire ou, plus généralement, de traumatisme oculaire impose une surveillance au long cours, dont le patient doit être prévenu.
Il est important de consigner, lors de l’examen initial, les éléments cliniques retrouvés, sans oublier l’œil adelphe. Les implications médicolégales sont toujours possibles dans ce contexte.
Le glaucome post-traumatique peut survenir des années après le traumatisme initial ; il est généralement unilatéral et associé à des signes évocateurs, retrouvés notamment à l’examen de l’angle iridocornéen (fig. 14-19).

Fig. 14-19 Mécanismes physiopathologiques pouvant intervenir dans le glaucome post-contusif.
Les glaucomes secondaires à angle ouvert liés au cristallin ou à la chirurgie du cristallin regroupent des entités différentes. Les glaucomes secondaires induits par le cristallin sont traités par ailleurs.
Une HTO généralement transitoire peut survenir au décours d’une chirurgie de cataracte sans complication peropératoire. Elle est souvent en rapport avec la présence résiduelle de produit viscoélastique et s’amende rapidement avec la prise d’un traitement hypotonisant oculaire. Une chirurgie compliquée de la cataracte peut être source d’HTO au décours immédiat de l’intervention.
Les implants de chambre antérieure à fixation angulaire sont souvent pourvoyeurs d’HTO et de glaucome à plus ou moins long terme [24]. Les implants à fixation irienne sont moins problématiques, mais néanmoins susceptibles d’entraîner une dispersion pigmentaire, voire une uvéite chronique pouvant déclencher une HTO.
L’implant de chambre postérieure susceptible de poser problème n’est pas celui positionné dans le sac capsulaire ; l’HTO survenant dans ce contexte est souvent liée à un implant positionné dans le sulcus (fig. 14-20). On peut alors observer un frottement de l’iris sur l’implant avec dispersion pigmentaire secondaire ; il peut également survenir une uvéite chronique du fait d’une interaction de l’implant avec les tissus uvéaux [18, 51].
L’ablation de l’implant responsable est souvent nécessaire ; malgré tout, l’HTO peut persister, imposant une prise en charge au long cours, voire la réalisation d’une chirurgie filtrante.

Fig. 14-20 Glaucome du pseudo-phake ; implant de chambre postérieure luxé. (Cliché : J.-P. Renard)
La majeure partie des glaucomes de l’aphake survient sur des yeux opérés de cataracte congénitale, dont la fréquence est d’environ 20 % à cinq ans [20, 25, 71, 88]. Le glaucome de l’aphake le plus fréquent est à angle ouvert [6] et peut survenir des années après la chirurgie initiale.
Les mécanismes physiopathologiques sont multiples : anomalies oculaires congénitales associées, inflammation chronique, maturation anormale du trabéculum, persistance de matériel cristallinien [19, 20, 59], interaction entre cellules trabéculaires et cellules épithéliales cristalliniennes [59], etc.
D’autres facteurs de risque ont été identifiés [4, 6, 17] : chirurgie réalisée pendant la première année de vie [39], petit diamètre cornéen [60, 81], antécédent familial de glaucome de l’aphake, capsulotomie ou vitrectomie dans le même temps opératoire. L’influence de la mise en place d’un implant demeure discutée comme facteur protecteur vis-à-vis de la survenue du glaucome [6, 17, 39].
Une surveillance à vie est nécessaire, l’incidence augmentant avec la durée de suivi [81] ; le pronostic de ce type de glaucome est en général réservé, et la prise en charge difficile.
Le glaucome par augmentation de la pression veineuse épisclérale est un glaucome secondaire à angle ouvert qui regroupe des étiologies multiples, souvent dans un contexte de pathologies générales associées.
Cette situation peut être aiguë ou devenir chronique avec parfois des conséquences sur la tête du nerf optique. Les signes cliniques sont parfois trompeurs et peu spécifiques, ou alors évocateurs en présence de veines épisclérales tortueuses et dilatées, d’une exophtalmie, de sang dans le canal de Schlemm visualisé en gonioscopie, associés à une HTO.
Les étiologies d’augmentation de la pression veineuse épisclérale peuvent être en rapport avec une obstruction veineuse, des anomalies artérioveineuses ou idiopathiques [67]. Une obstruction veineuse pourra être rapprochée d’une orbitopathie dysthyroïdienne, d’un syndrome de la veine cave supérieure, d’une thrombose du sinus caverneux ou d’un processus occupant l’espace rétrobulbaire. Les anomalies artérioveineuses susceptibles d’induire une hyperpression veineuse épisclérale sont les fistules carotidocaverneuses post-traumatiques ou spontanées, avec leur cortège de signes cliniques plus ou moins bruyants, les varices orbitaires et le syndrome de Sturge-Weber.
La prise en charge comportera idéalement un traitement étiologique, qui permettra d’optimiser le traitement de l’HTO. Cette dernière peut nécessiter l’instauration d’un traitement médical, voire chirurgical ; notons la moindre efficacité des analogues de prostaglandines ainsi que des trabéculoplasties dans les stades aigus. En cas d’indication chirurgicale, il est sans doute important de ne pas méconnaître les risques d’hémorragie choroïdienne, d’effusion uvéale et d’hypotonie importante.
D’autres situations peuvent éventuellement générer ou être associées à des glaucomes à angle ouvert :
certaines pathologies comme le syndrome de Stickler systémique héréditaire de dégénérescence vitréorétinienne [84] ou le syndrome de Schwartz dans lequel on observe une HTO compliquant un décollement de rétine prolongé par encombrement trabéculaire par les segments externes des photorécepteurs [77, 79] ;
le glaucome à cellules fantômes dans le contexte d’une hémorragie intravitréenne ;
dans les suites d’une chirurgie vitréorétinienne, en particulier avec l’utilisation de certains tamponnements comme des gaz expansifs ou l’huile de silicone ;
certaines tumeurs intra-oculaires, en particulier celles situées dans le segment antérieur ou dans le corps ciliaire, par invasion de l’angle iridocornéen ou par infiltration carcinomateuse.
Retenir
Le glaucome primitif à angle ouvert est un diagnostic d’élimination ; les glaucomes secondaires sont à évoquer en prérequis, afin d’en ajuster la prise en charge.
Les glaucomes uvéitique et cortico-induit imposent rigueur et vigilance quant au suivi des patients concernés. La prise en charge du phénomène inflammatoire passe aussi par la gestion de son étiologie.
Le glaucome post-traumatique est une entité à évoquer au stade aigu d’un traumatisme pour optimiser la gestion des modifications anatomiques induites, mais également au stade tardif, sans limitation dans le temps, durant la surveillance d’un œil traumatisé.
Le glaucome de l’aphake est relativement fréquent dans le contexte d’une cataracte congénitale opérée ; l’identifier est fondamental pour limiter l’évolution parfois compliquée de ce type de glaucome.
[1] Anand N. Deep sclerectomy with mitomycin C for glaucoma secondary to uveitis. Eur J Ophthalmol. 2011 ; 21 : 708-14.
[2] Armaly MF. Effect of corticosteroids on intraocular pressure and fluid dynamics. I. The effect of dexamethasone in the normal eye. Arch Ophthalmol. 1963 ; 70 : 482-91.
[3] Armaly MF. Effect of corticosteroids on intraocular pressure and fluid dynamics. II. The effect of dexamethasone in the glaucomatous eye. Arch Ophthalmol. 1963 ; 70 : 492-9.
[4] Asrani SG, Wilensky JT. Glaucoma after congenital cataract surgery. Ophthalmology. 1995 ; 102 : 863-7.
[5] Ataullah S, Biswas S, Artes PH, et al. Long term results of diode laser cycloablation in complex glaucoma using the Zeiss Visulas II system. Br J Ophthalmol. 2002 ; 86 : 39-42.
[6] Beck AD, Freedman SF, Lynn MJ, et al. Glaucoma-related adverse events in the Infant Aphakia Treatment Study : 1-year results. Arch Ophthalmol. 2012 ; 130 : 300-5.
[7] Becker B. Diabetes mellitus and primary open-angle glaucoma. The XXVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 1971 ; 71 : 1-16.
[8] Becker B. Intraocular pressure response to topical corticosteroids. Invest Ophthalmol. 1965 ; 4 : 198-205.
[9] Becker B, Hahn KA. Topical corticosteroids and heredity in primary open-angle glaucoma. Am J Ophthalmol. 1964 ; 57 : 543-51.
[10] Becker B, Mills DW. Corticosteroids and Intraocular Pressure. Arch Ophthalmol. 1963 ; 70 : 500-7.
[11] Bollinger K, Kim J, Lowder CY, et al. Intraocular pressure outcome of patients with fluocinolone acetonide intravitreal implant for noninfectious uveitis. Ophthalmology. 2011 ; 118 : 1927-31.
[12] Broadway DC, Bates AK, Lightman SL, et al. The importance of cellular changes in the conjunctiva of patients with uveitic glaucoma undergoing trabeculectomy. Eye. 1993 ; 7 : 495-501.
[13] Campbell D. Traumatic glaucoma. In : Shingleton B, Kenyon K (eds). Eye trauma. St. Louis, Mosby Year Book, 1991 : 117-25.
[14] Cantrill HL, Palmberg PF, Zink HA, et al. Comparison of in vitro potency of corticosteroids with ability to raise intraocular pressure. Am J Ophthalmol. 1975 ; 79 : 1012-7.
[15] Ceballos EM, Beck AD, Lynn MJ. Trabeculectomy with antiproliferative agents in uveitic glaucoma. J Glaucoma. 2002 ; 11 : 189-96.
[16] Ceballos EM, Parrish RK, Schiffman JC. Outcome of Baerveldt glaucoma drainage implants for the treatment of uveitic glaucoma. Ophthalmology. 2002 ; 109 : 2256-60.
[17] Chak M, Rahi JS. British Congenital Cataract Interest G. Incidence of and factors associated with glaucoma after surgery for congenital cataract : findings from the British Congenital Cataract Study. Ophthalmology. 2008 ; 115 : 1013-8.
[18] Chang DF, Masket S, Miller KM, et al. Complications of sulcus placement of single-piece acrylic intraocular lenses : recommendations for backup IOL implantation following posterior capsule rupture. J Cataract Refractive Surg. 2009 ; 35 : 1445-58.
[19] Chen TC, Bhatia LS, Halpern EF, Walton DS. Risk factors for the development of aphakic glaucoma after congenital cataract surgery. Journal of Pediatric Ophthalmology and Strabismus. 2006 ; 43 : 274-80 ; quiz : 306-7.
[20] Chen TC, Walton DS, Bhatia LS. Aphakic glaucoma after congenital cataract surgery. Arch Ophthalmol. 2004 ; 122 : 1819-25.
[21] Clark AF, Wilson K, McCartney MD, et al. Glucocorticoid-induced formation of cross-linked actin networks in cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 1994 ; 35 : 281-94.
[22] Clarke LA, Guex-Crosier Y, Hofer M. Epidemiology of uveitis in children over a 10-year period. Clin Exp Rheumatol. 2013 ; 31 : 633-7.
[23] Cochereau I. [Glaucoma and uveitis]. J Fr Ophtalmol. 2003 ; 26 (n° spécial 2) : S10-2.
[24] Collins JF, Gaster RN, Krol WF, et al. A comparison of anterior chamber and posterior chamber intraocular lenses after vitreous presentation during cataract surgery : the Department of Veterans Affairs Cooperative Cataract Study. Am J Ophthalmol. 2003 ; 136 : 1-9.
[25] Comer RM, Kim P, Cline R, Lyons CJ. Cataract surgery in the first year of life : aphakic glaucoma and visual outcomes. Can J Ophthalmol. 2011 ; 46 : 148-52.
[26] Crouch ER, Crouch ER. Management of traumatic hyphema : therapeutic options. Journal of Pediatric Ophthalmology and Strabismus. 1999 ; 36 : 238-50 ; quiz : 79-80.
[27] Crouch ER, Williams PB, Gray MK, et al. Topical aminocaproic acid in the treatment of traumatic hyphema. Arch Ophthalmol. 1997 ; 115 : 1106-12.
[28] Dannenberg AL, Parver LM, Brechner RJ, Khoo L. Penetration eye injuries in the workplace. The National Eye Trauma System Registry. Arch Ophthalmol. 1992 ; 110 : 843-8.
[29] Deans R, Noel LP, Clarke WN. Oral administration of tranexamic acid in the management of traumatic hyphema in children. Can J Ophthalmol. 1992 ; 27 : 181-3.
[30] Ducasse A, Denis P. [Good practices in traumatic glaucoma]. J Fr Ophtalmol. 2000 ; 23 : 295-8.
[31] Dupas B, Fardeau C, Cassoux N, et al. Deep sclerectomy and trabeculectomy in uveitic glaucoma. Eye. 2010 ; 24 : 310-4.
[32] Espildora J, Vicuna P, Diaz E. [Cortisone-induced glaucoma : a report on 44 affected eyes (author’s transl)]. J Fr Ophtalmol. 1981 ; 4 : 503-8.
[33] Fingert JH, Alward WL, Wang K, et al. Assessment of SNPs associated with the human glucocorticoid receptor in primary open-angle glaucoma and steroid responders. Mol Vis. 2010 ; 16 : 596-601.
[34] Foster CS, Havrlikova K, Baltatzis S, et al. Secondary glaucoma in patients with juvenile rheumatoid arthritis-associated iridocyclitis. Acta Ophthalmol Scand. 2000 ; 78 : 576-9.
[35] Francois J. Corticosteroid glaucoma. Ann Ophthalmol. 1977 ; 9 : 1075-80.
[36] Gaston H, Absolon MJ, Thurtle OA, Sattar MA. Steroid responsiveness in connective tissue diseases. Br J Ophthalmol. 1983 ; 67 : 487-90.
[37] Gharaibeh A, Savage HI, Scherer RW, et al. Medical interventions for traumatic hyphema. Cochrane Database of Systematic Reviews. 2011 ; 1 : CD005431.
[38] Gillies MC, Sutter FK, Simpson JM, et al. Intravitreal triamcinolone for refractory diabetic macular edema : two-year results of a double-masked, placebo-controlled, randomized clinical trial. Ophthalmology. 2006 ; 113 : 1533-8.
[39] Haargaard B, Ritz C, Oudin A, et al. Risk of glaucoma after pediatric cataract surgery. Invest Ophthalmol Vis Sci. 2008 ; 49 : 1791-6.
[40] Heiligenhaus A, Niewerth M, Ganser G, et al. German Uveitis in Childhood Study G. Prevalence and complications of uveitis in juvenile idiopathic arthritis in a population-based nation-wide study in Germany : suggested modification of the current screening guidelines. Rheumatology. 2007 ; 46 : 1015-9.
[41 ]Heinz C, Koch JM, Zurek-Imhoff B, Heiligenhaus A. Prevalence of uveitic secondary glaucoma and success of nonsurgical treatment in adults and children in a tertiary referral center. Ocular Immunology and Inflammation. 2009 ; 17 : 243-8.
[42] Herbert HM, Viswanathan A, Jackson H, Lightman SL. Risk factors for elevated intraocular pressure in uveitis. J Glaucoma. 2004 ; 13 : 96-9.
[43] Herschler J. Increased intraocular pressure induced by repository corticosteroids. Am J Ophthalmol. 1976 ; 82 : 90-3.
[44] Jap A, Chee SP. Viral anterior uveitis. Curr Opin Ophthalmol. 2011 ; 22 : 483-8.
[45] Jonas JB, Degenring RF, Kreissig I, et al. Intraocular pressure elevation after intravitreal triamcinolone acetonide injection. Ophthalmology. 2005 ; 112 : 593-8.
[46] Jones NP. Fuchs’ heterochromic uveitis : a reappraisal of the clinical spectrum. Eye. 1991 ; 5 : 649-61.
[47] Jones NP. Glaucoma in Fuchs’ heterochromic uveitis : aetiology, management and outcome. Eye. 1991 ; 5 : 662-7.
[48] Kaufman JH, Tolpin DW. Glaucoma after traumatic angle recession. A ten-year prospective study. Am J Ophthalmol. 1974 ; 78 : 648-54.
[49] Kersey JP, Broadway DC. Corticosteroid-induced glaucoma : a review of the literature. Eye. 2006 ; 20 : 407-16.
[50] Kiddee W, Trope GE, Sheng L, et al. Intraocular pressure monitoring post-intravitreal steroids : a systematic review. Surv Ophthalmol. 2013 ; 58 : 291-310.
[51] Kirk KR, Werner L, Jaber R, et al. Pathologic assessment of complications with asymmetric or sulcus fixation of square-edged hydrophobic acrylic intraocular lenses. Ophthalmology. 2012 ; 119 : 907-13.
[52] Kishi A, Nao-i N, Sawada A. Ultrasound biomicroscopic findings of acute angle-closure glaucoma in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1996 ; 122 : 735-7.
[53] Labetoulle M, Colin J. [Current concepts in the treatment of herpetic keratitis]. J Fr Ophtalmol. 2012 ; 35 : 292-307.
[54] Lean M. Use of ACTH and cortisone. Transactions of the American Ophthalmological Society. 1950 ; 48 : 293-6.
[55] Manners T, Salmon JF, Barron A, et al. Trabeculectomy with mitomycin C in the treatment of post-traumatic angle recession glaucoma. Br J Ophthalmol. 2001 ; 85 : 159-63.
[56] Merayo-Lloves J, Power WJ, Rodriguez A, et al. Secondary glaucoma in patients with uveitis. Ophthalmologica. 1999 ; 213 : 300-4.
[57] Mermoud A, Salmon JF, Barron A, et al. Surgical management of post-traumatic angle recession glaucoma. Ophthalmology. 1993 ; 100 : 634-42.
[58] Mermoud A, Salmon JF, Straker C, Murray AD. Post-traumatic angle recession glaucoma : a risk factor for bleb failure after trabeculectomy. Br J Ophthalmol. 1993 ; 77 : 631-4.
[59] Michael I, Shmoish M, Walton DS, Levenberg S. Interactions between trabecular meshwork cells and lens epithelial cells : a possible mechanism in infantile aphakic glaucoma. Invest Ophthalmol Vis Sci. 2008 ; 49 : 3981-7.
[60] Mills MD, Robb RM. Glaucoma following childhood cataract surgery. Journal of Pediatric Ophthalmology and Strabismus. 1994 ; 31 : 355-60 ; discussion : 61.
[61] Molteno AC, Sayawat N, Herbison P. Otago glaucoma surgery outcome study : long-term results of uveitis with secondary glaucoma drained by Molteno implants. Ophthalmology. 2001 ; 108 : 605-13.
[62] Moorthy RS, Mermoud A, Baerveldt G, et al. Glaucoma associated with uveitis. Surv Ophthalmol. 1997 ; 41 : 361-94.
[63] Neri P, Azuara-Blanco A, Forrester JV. Incidence of glaucoma in patients with uveitis. J Glaucoma. 2004 ; 13 : 461-5.
[64] Nguyen TD, Chen P, Huang WD, et al. Gene structure and properties of TIGR, an olfactomedin-related glycoprotein cloned from glucocorticoid-induced trabecular meshwork cells. J Biol Chem. 1998 ; 273 : 6341-50.
[65] Panek WC, Holland GN, Lee DA, Christensen RE. Glaucoma in patients with uveitis. Br J Ophthalmol. 1990 ; 74 : 223-7.
[66] Paterson CA, Pfister RR. Intraocular pressure changes after alkali burns. Arch Ophthalmol. 1974 ; 91 : 211-8.
[67] Piltz-Seymour J, Uhler TA. Others secondary glaucomas. In : Shaarawy T et al. (eds). Glaucoma (volume 1). Elsevier, 2009 : 419-30.
[68] Podos SM, Becker B, Morton WR. High myopia and primary open-angle glaucoma. Am J Ophthalmol. 1966 ; 62 : 1038-43.
[69] Pogorzalek N, de Monchy I, Gendron G, Labetoulle M. [Hypertony and uveitis : 103 cases of uveitis]. J Fr Ophtalmol. 2011 ; 34 : 157-63.
[70] Qian Y, Acharya NR. Juvenile idiopathic arthritis-associated uveitis. Curr Opin Ophthalmol. 2010 ; 21 : 468-72.
[71] Rabiah PK. Frequency and predictors of glaucoma after pediatric cataract surgery. Am J Ophthalmol. 2004 ; 137 : 30-7.
[72] Rao PV, Deng PF, Kumar J, Epstein DL. Modulation of aqueous humor outflow facility by the Rho kinase-specific inhibitor Y-27632. Invest Ophthalmol Vis Sci. 2001 ; 42 : 1029-37.
[73] Renard JP. Glaucomes post-traumatiques. In : Chirurgie des glaucomes. Bulletin des Sociétés d’Ophtalmologie de France, éditions Lamy, 2005 : 325-33.
[74] Rothova A, Suttorp-van Schulten MS, Frits Treffers W, Kijlstra A. Causes and frequency of blindness in patients with intraocular inflammatory disease. Br J Ophthalmol. 1996 ; 80 : 332-6.
[75] Rubin B, Taglienti A, Rothman RF, et al. The effect of selective laser trabeculoplasty on intraocular pressure in patients with intravitreal steroid-induced elevated intraocular pressure. J Glaucoma. 2008 ; 17 : 287-92.
[76] Salmon JF, Mermoud A, Ivey A, et al. The detection of post-traumatic angle recession by gonioscopy in a population-based glaucoma survey. Ophthalmology. 1994 ; 101 : 1844-50.
[77] Schwartz A. Chronic open-angle glaucoma secondary to rhegmatogenous retinal detachment. Am J Ophthalmol. 1973 ; 75 : 205-11.
[78] Shazly TA, Aljajeh M, Latina MA. Posner-Schlossman glaucomatocyclitic crisis. Semin Ophthalmol. 2011 ; 26 : 282-4.
[79] Sellem E. Glaucome et maladies rétiniennes et vitréorétiniennes. In : Schnyder C, Mermoud A (eds). Glaucome. Issy-les-Moulineaux, Elsevier, 2005 : 294-301.
[80] Siddique SS, Suelves AM, Baheti U, Foster CS. Glaucoma and uveitis. Surv Ophthalmol. 2013 ; 58 : 1-10.
[81] Simon JW, Mehta N, Simmons ST, et al. Glaucoma after pediatric lensectomy/vitrectomy. Ophthalmology. 1991 ; 98 : 670-4.
[82] Snyder RW, Stamer WD, Kramer TR, Seftor RE. Corticosteroid treatment and trabecular meshwork proteases in cell and organ culture supernatants. Exper Eye Res. 1993 ; 57 : 461-8.
[83] Spaeth GL. Traumatic hyphema, angle recession, dexaméthasone hypertension, and glaucoma. Arch Ophthalmol. 1967 ; 78 : 714-21.
[84] Stickler GB, Belau PG, Farrell FJ, et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clinic Proceedings. 1965 ; 40 : 433-55.
[85] Suttorp-Schulten MS, Rothova A. The possible impact of uveitis in blindness : a literature survey. Br J Ophthalmol. 1996 ; 80 : 844-8.
[86] Tektas OY, Heinz C, Heiligenhaus A, et al. Morphological changes of trabeculectomy specimens in different kinds of uveitic glaucoma. Curr Eye Res. 2011 ; 36 : 442-8.
[87] Tesluk GC, Spaeth GL. The occurrence of primary open-angle glaucoma in the fellow eye of patients with unilateral angle-cleavage glaucoma. Ophthalmology. 1985 ; 92 : 904-11.
[88] Vishwanath M, Cheong-Leen R, Taylor D, et al. Is early surgery for congenital cataract a risk factor for glaucoma ? Br J Ophthalmol. 2004 ; 88 : 905-10.
[89] Volpe NJ, Larrison WI, Hersh PS, et al. Secondary hemorrhage in traumatic hyphema. Am J Ophthalmol. 1991 ; 112 : 507-13.
[90] Warwar RE, Bullock JD, Ballal D. Cystoid macular edema and anterior uveitis associated with latanoprost use. Experience and incidence in a retrospective review of 94 patients. Ophthalmology. 1998 ; 105 : 263-8.
[91] Weinreb RN, Polansky JR, Kramer SG, Baxter JD. Acute effects of dexamethasone on intraocular pressure in glaucoma. Invest Ophthalmol Vis Sci. 1985 ; 26 : 170-5.
[92] Wordinger RJ, Clark AF. Effects of glucocorticoids on the trabecular meshwork : towards a better understanding of glaucoma. Progress in Retinal and Eye Research. 1999 ; 18 : 629-67.
[93] Yue BY. The extracellular matrix and its modulation in the trabecular meshwork. Surv Ophthalmol. 1996 ; 40 : 379-90.
A. Bastelica
Les mécanismes aboutissant à un glaucome chronique par fermeture primitive de l’angle sont souvent multifactoriels : blocage pupillaire, iris plateau, goniosynéchies.
La gonioscopie est indispensable, souvent appuyée par l’imagerie du segment antérieur.
La réalisation d’une iridotomie périphérique est systématique.
Une iridoplastie, une phaco-exérèse et/ou une chirurgie filtrante peuvent être ultérieurement envisagées en cas de persistance d’une hypertension intra-oculaire (HTO).
La différenciation entre glaucome primitif à angle ouvert (GPAO) et glaucome chronique par fermeture primitive de l’angle (GCFPA) est essentielle. Cette dernière affection est potentiellement plus grave et sa prise en charge thérapeutique est différente : l’iridotomie au laser est en effet la pierre angulaire du traitement du GCFPA alors qu’elle n’est pas indiquée dans le GPAO.
Par ailleurs, la chirurgie filtrante comporte plus de risques de complications postopératoires graves sur ces yeux (effusion choroïdienne, hémorragie expulsive, glaucome malin).
L’extraction du cristallin par phaco-émulsification est une approche plus récente de la gestion de ces patients et suscite un intérêt croissant parmi les spécialistes internationaux.
L’absence de cause décelable définit le caractère primitif de la fermeture de l’angle (FPA). La fermeture de l’angle résultant d’une pathologie oculaire (uvéite, néovascularisation, etc.) ou de l’action de forces s’exerçant au niveau du cristallin ou en arrière de lui (cataracte, glaucome malin, etc.) est classée dans les formes secondaires.
La terminologie et la définition du GCFPA ont été précisées récemment par Foster et al. [4] qui distinguent trois stades physiopathologiques (tableau 14-2) :
suspicion de fermeture de l’angle. Il s’agit d’une prédisposition anatomique de l’angle iridocornéen à la fermeture. Un contact entre la racine de l’iris et le trabéculum postérieur est possible (« occludable angle »). En gonioscopie, ce type d’angle est caractérisé par l’impossibilité de visualiser le trabéculum postérieur (stade 0 à 2 de la classification de Shaffer) sur plus de 270°. La pression intra-oculaire (PIO) est normale, il n’y a pas de synéchie antérieure périphérique (SAP), l’aspect du disque optique et le champ visuel sont normaux. Savoir si les angles avec un contact sur 180° doivent être inclus dans cette catégorie est encore débattu ;
fermeture de l’angle. Au stade précédent s’associe une élévation de la PIO et/ou associée à des SAP. L’aspect du disque optique et le champ visuel sont normaux ;
glaucome par fermeture de l’angle. Au stade précédent s’associent des signes de neuropathie optique glaucomateuse (disque optique et/ou champ visuel).
Cette classification consensuelle est actuellement admise. Son défaut est qu’elle ne tient pas compte du mécanisme de fermeture.

Tableau 14.2 – Classification des fermetures primitives de l’angle selon Foster [5].
Les perspectives pour 2020 font état de 79,6 millions de glaucomes dans le monde, dont 21 millions de GCFPA [21].
Le GCFPA est une affection particulièrement grave : 25 % des patients atteints seront aveugles, et le GCFPA sera responsable de la moitié des cécités dues au glaucome, soit 5,3 millions [19, 26].
Le facteur de risque oculaire le plus important est la présence d’un segment antérieur de petite dimension, et la profondeur de la chambre antérieure est d’ailleurs fortement corrélée à la fermeture de l’angle et au GCFPA [5].
Les yeux à risque présentent :
un segment antérieur étroit avec une petite longueur axiale, un petit diamètre et un petit rayon de courbure de la cornée ;
une augmentation de volume et une position antérieure du cristallin.
Les facteurs de risque démographiques sont l’âge, le sexe féminin, l’origine asiatique et l’existence d’antécédents familiaux (tableau 14-3).

Tableau 14.3 – Facteurs de risque de fermeture de l’angle.
L’origine des forces provoquant un blocage du trabéculum par l’iris peut se situer à quatre niveaux anatomiques : l’iris (niveau 1, bloc pupillaire), le corps ciliaire (niveau 2, iris plateau), le cristallin (niveau 3, glaucome phacomorphique) et en arrière du cristallin (niveau 4, glaucome malin). Ces deux dernières formes de glaucome sont classées dans les glaucomes secondaires par fermeture de l’angle.
Le bloc pupillaire est le mécanisme prédominant dans 75 % des cas de fermeture primitive de l’angle [6].
Le bloc pupillaire relatif est une exagération du phénomène physiologique dans lequel le flux de l’humeur aqueuse est ralenti lors du passage transpupillaire par le contact iridolenticulaire [23]. L’humeur aqueuse s’accumule dans la chambre postérieure provoquant un gradient de pression entre chambres postérieure et antérieure. La racine de l’iris se déplace vers l’avant et entre en contact avec le trabéculum et/ou la cornée périphérique, provoquant un obstacle prétrabéculaire.
Le bloc pupillaire nécessite une prédisposition anatomique : les yeux à risque ont une chambre antérieure étroite. Cependant, il apparaît de plus en plus probable que l’anatomie n’explique pas tout : ces yeux doivent avoir par ailleurs un comportement physiologique anormal. Cette hypothèse repose sur plusieurs constatations :
la plupart des individus avec petits yeux et angles étroits ne développeront jamais la maladie. Seul un angle étroit sur dix présentera une fermeture de l’angle ;
la simple observation statique par gonioscopie ou UBM n’est pas capable d’identifier clairement les personnes à risque de GCFPA ;
la population chinoise présente cinq fois plus de risque de GCFPA que les populations européenne et africaine, alors qu’aucune différence dans la longueur axiale ou la profondeur de la chambre antérieure, entre ces populations, n’a pu être mise en évidence [3].
Des travaux récents sont venus confirmer cette hypothèse d’anomalie dynamique de l’iris : l’OCT a permis de mettre en évidence, chez les sujets normaux, un amincissement de la périphérie de l’iris lors de la dilatation pupillaire. Ce phénomène pourrait s’expliquer par une issue de fluide hors du stroma irien vers la chambre antérieure. L’iris des sujets présentant une fermeture de l’angle semble avoir une faible conductibilité aux fluides de sorte que, lors de la dilatation, l’amincissement de l’iris ne se produit pas, ce dernier pouvant même augmenter de volume [1, 20, 22].
Le rôle de l’expansion choroïdienne a également été évoqué.
Ce mécanisme de fermeture correspond à un blocage antérieur sans blocage pupillaire. La chambre antérieure est de profondeur habituellement normale au centre.
Les sujets sont généralement plus jeunes (30-50 ans), moins hypermétropes et ont souvent des antécédents familiaux.
Le mécanisme d’iris plateau est souvent méconnu et donc largement sous-estimé. Certes, la forme isolée est rare, mais les formes mixtes sont fréquentes, un bloc pupillaire s’associant souvent à un certain degré d’iris plateau. Cela explique que, dans la plupart des cas, une iridectomie périphérique permet une ouverture de l’angle suffisante mais, chez certains patients, une apposition iridotrabéculaire persiste malgré tout après une iridotomie [10, 13]. En réalité, il faut maintenant distinguer la « configuration iris plateau » du « syndrome iris plateau ».
La configuration iris plateau est la résultante de variations dans l’anatomie de l’iris et du corps ciliaire qui mettent en contact la périphérie irienne avec le trabéculum :
insertion antérieure de l’iris sur le corps ciliaire ;
angulation anormale de la racine de l’iris ;
surface de l’iris plane ;
iris épais en périphérie (bourrelet) ;
antéposition du corps ciliaire, qui pousse la racine de l’iris vers l’avant, avec réduction, voire disparition, du sulcus ciliaire [17].
Le syndrome iris plateau est de description plus récente. Il est plus rare et se réfère à l’état dans lequel l’angle iridocornéen peut se fermer, soit spontanément, soit pharmacologiquement, après qu’une iridotomie ait éliminé tout élément de bloc pupillaire.
Elles font suite à une apposition iridotrabéculaire prolongée. Souvent précédées par une pigmentation trabéculaire, elles débutent habituellement dans le quadrant supérieur. Elles peuvent se constituer de deux manières : soit de l’anneau de Schwalbe vers le fond de l’angle, soit à partir du fond de l’angle vers la périphérie de la cornée réalisant la forme rampante ou « creeping glaucoma ». Cette forme, insidieuse, est particulièrement fréquente en cas d’iris épais (sujets asiatiques).
La fermeture devient pathologique lorsqu’au contact iridotrabéculaire s’associe une élévation de la PIO ou des SAP. L’hypertonie oculaire peut alors être responsable de dommages structuraux au niveau de la tête du nerf optique.
L’apposition iridotrabéculaire va diminuer le flux trabéculaire d’humeur aqueuse par différents mécanismes se succédant au cours de l’évolution de la maladie :
obstruction prétrabéculaire réversible ;
altérations du trabéculum lui-même. Les frottements répétés de l’iris sur le trabéculum sont responsables d’une altération anatomique et fonctionnelle du filtre trabéculaire. Il s’agit d’une accumulation de pigments, d’une dégénérescence cellulaire non inflammatoire, d’une perte de cellules endothéliales et d’une altération de l’architecture trabéculaire [8] ;
la constitution de SAP est le mécanisme de diminution de la facilité de l’écoulement d’humeur aqueuse le plus évident et il est unanimement accepté. Des dommages irréversibles se produisent au niveau du trabéculum dans les zones de persistance de SAP, avec prolifération de tissu irien dans les espaces intertrabéculaires. Le degré d’élévation de la PIO est associé à l’étendue des synéchies [9].
L’établissement du diagnostic et l’analyse du ou, plus souvent, des mécanismes de fermeture de l’angle sont primordiaux. Ils permettront de choisir la stratégie thérapeutique la mieux adaptée à chaque cas afin d’éviter la progression de la neuropathie optique glaucomateuse et l’atteinte fonctionnelle grave qui peut en découler.
L’évaluation de la profondeur de la chambre antérieure et l’appréciation du degré d’ouverture de l’angle sont les deux critères fondamentaux.
L’examen biomicroscopique permet d’apprécier la profondeur de la chambre antérieure. Une faible profondeur de chambre antérieure mesurée au centre prédispose à la fermeture de l’angle. Le risque est de 100 % pour une profondeur de chambre antérieure inférieure à 1,3 mm et seulement de 1 % si elle est supérieure à 2,2 mm. De même, le risque de fermeture est réel si la profondeur de la chambre antérieure en périphérie, nasale ou temporale, est inférieure au quart de l’épaisseur cornéenne (signe de Van Herick).
La gonioscopie demeure l’examen clé. Elle doit être pratiquée au moindre doute chez tout patient glaucomateux, suspect de l’être ou à risque (HTO). Elle précisera le degré d’ouverture de l’angle, le site d’insertion de l’iris, la courbure périphérique de l’iris (convexe ou concave), les zones d’apposition iridotrabéculaire et le degré de pigmentation du trabéculum. Cet examen statique sera complété, en cas de non-visibilité de l’angle, par un examen dynamique. L’utilisation d’un verre de petit diamètre (Posner, Sussman, etc.) est nécessaire pour produire une indentation du centre de la cornée. Cette indentation, en provoquant une chasse de l’humeur aqueuse en périphérie, va permettre de préciser le caractère réversible ou non de la fermeture ainsi que le mécanisme en cause :
si, lors de l’indentation, l’angle s’ouvre et les différents éléments deviennent visibles, il s’agit d’une simple apposition iridotrabéculaire (réversible). Le comportement de l’iris permettra de définir le mécanisme en cause. Si l’iris devient concave, il s’agit d’un bloc pupillaire. En revanche, si l’angle s’ouvre plus difficilement et si l’iris prend un aspect « en double bosse », il s’agit d’un iris plateau. Quand le cristallin est impliqué dans la fermeture, l’iris se moule sur la face antérieure du cristallin et demeure convexe avec de petits mouvements postérieurs ;
si, lors de l’indentation, l’angle demeure fermé, la fermeture est irréversible du fait de la présence de SAP et l’examen en précisera l’étendue.
Elle a un intérêt certain dans l’évaluation des sujets présentant un angle étroit, mais ne remplace pas l’examen gonioscopique.
La tomographie à cohérence optique (OCT) du segment antérieur permet une bonne appréciation de l’ouverture de l’angle ainsi qu’un examen dynamique. Elle peut parfois révéler une fermeture de l’angle intermittente (test à l’obscurité). La biomicroscopie ultrasonique (UBM) présente un intérêt majeur dans le diagnostic d’iris plateau et pour préciser la position plus ou moins antérieure du corps ciliaire [17] (eFig. 14-2).
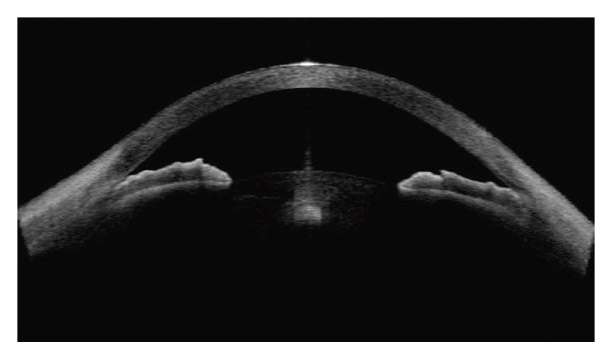
eFig. 14-2 Fermeture de l’angle en OCT. (Cliché : J. Laloum.)
Le GCFPA peut se présenter sous forme aiguë, subaiguë ou chronique.
La forme aiguë pose peu de problèmes diagnostiques.
La forme intermittente est difficile à diagnostiquer car les épisodes de fermeture se résolvent spontanément, sans laisser de séquelles sinon des dépôts pigmentés irréguliers dans l’angle témoignant du contact iridocornéen périphérique.
La forme chronique se développe progressivement, de manière insidieuse. Sa fréquence est probablement sous-estimée. Le diagnostic est souvent confondu avec un glaucome à angle ouvert. Elle se caractérise par la présence d’une neuropathie glaucomateuse associée à des goniosynéchies.
Les atteintes du nerf optique et du champ visuel sont identiques dans le GCFPA et le GPAO [2].
Parfois, en cas d’épisode de forte et brève HTO, la papille peut être peu excavée et la pâleur prédominer, mimant alors une neuropathie non glaucomateuse.
Le GCFPA est associé à un fort risque de perte visuelle : il est cinq fois plus responsable de cécité que le glaucome à angle ouvert.
Le traitement a deux objectifs : traiter l’HTO et ouvrir l’angle, afin d’éviter la progression de la neuropathie glaucomateuse et de prévenir la survenue d’une crise de glaucome aigu.
Pour abaisser la PIO, un traitement médical doit être institué en première intention. Toutes les classes médicamenteuses peuvent être utilisées. La pilocarpine conserve son intérêt sur ce terrain pour son action sur l’angle iridocornéen.
Toutefois, l’action thérapeutique essentielle est dirigée sur l’iris ou sur le cristallin.
L’iridectomie permet de lever le bloc pupillaire et d’ouvrir l’angle iridocornéen [11]. Elle se pratique habituellement au laser Nd-YAG ou à l’argon, exceptionnellement de manière chirurgicale.
L’iridoplastie (ou iridorétraction périphérique, ou gonioplastie) au laser à l’argon est indiquée si l’angle demeure fermé malgré la présence d’une iridotomie fonctionnelle, et en l’absence de SAP étendues. Elle est particulièrement indiquée en cas d’iris plateau (eFig. 14-3).
La phaco-exérèse a la capacité d’approfondir la chambre antérieure et d’élargir l’angle en agissant sur les différents mécanismes de fermeture : bloc pupillaire, volume et position du cristallin [7, 9, 18]. Elle a récemment suscité de l’intérêt chez les spécialistes internationaux du fait de sa capacité à diminuer la PIO, souvent de manière importante, sur les yeux avec angle étroit [12, 14, 24, 25]. Dans certaines études, malgré des PIO élevées en préopératoire, deux tiers des patients pouvaient se passer de traitement après ablation du cristallin seule.
La trabéculectomie perforante est la technique de référence pour abaisser la PIO dans le GCFPA. La chirurgie non perforante est en principe contre-indiquée sur ce terrain. Les antimétabolites sont souvent indiqués sur ces yeux.
La chirurgie est plus risquée sur ces yeux que dans le GPAO et expose à des complications parfois dramatiques tels que le glaucome malin, l’effusion uvéale, voire l’hémorragie suprachoroïdienne.
La phaco-émulsification est toutefois plus délicate dans ce contexte (mauvaise dilatation pupillaire, cristallin volumineux, zonule fragile, poussée vitréenne). Les risques de complications per- et postopératoires sont plus importants. On peut citer :
les pics d’HTO potentiellement dangereux sur des yeux avec déficit sévère du champ visuel ;
le glaucome malin ;
la décompensation cornéenne endothéliale ;
les problèmes liés à l’implant : erreur de calcul [17], tilt et décentrement, plus fréquents.
Les chirurgies combinées peuvent associer à la phaco-exérèse :
une libération des synéchies angulaires (goniosynéchiolyse). Elle est efficace chez les patients dont les SAP sont récentes – moins de six mois à un an ;
une chirurgie filtrante. Cette chirurgie combinée peut être effectuée en un temps (phacotrabéculectomie) ou de manière séquentielle. Les résultats pressionnels de la phacotrabéculectomie sont comparables à ceux de la trabéculectomie seule. Les complications chirurgicales sont plus fréquentes que lorsque la phaco-émulsification est pratiquée seule. Une technique rigoureuse [15] et la pratique d’une chirurgie non perforante, si l’état de l’angle l’autorise, permettent de réduire la fréquence des complications dans la chirurgie combinée.
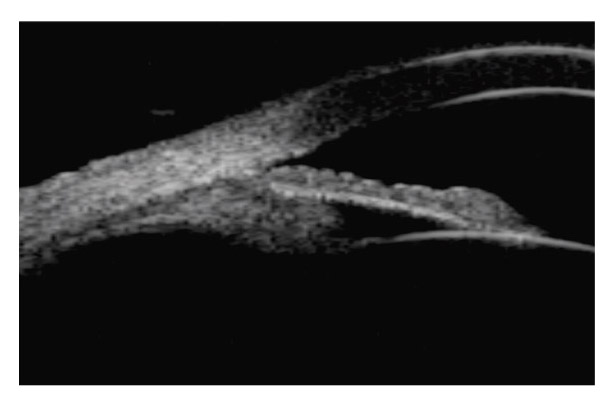
eFig. 14-3 UBM iris plateau. (Cliché : O. Bergès.)
Il n’y a pas de consensus concernant la meilleure stratégie chirurgicale dans le GCFPA. La prise en charge standard actuelle est une approche progressive consistant en une combinaison de laser, de traitement médical et de chirurgie. L’iridotomie bilatérale au laser est le traitement initial recommandé. Ensuite, le choix thérapeutique s’appuiera sur plusieurs considérations :
le mécanisme sous-jacent (bloc pupillaire, iris plateau) ;
le stade de la maladie (présence et sévérité de la neuropathie optique glaucomateuse) ;
l’existence de dégâts trabéculaires importants : HTO et extension des SAP en gonioscopie ;
le niveau de la PIO avant traitement ;
le traitement médical en cours : nombre et nature des traitements, effets secondaires, observance ;
le degré de cataracte.
Au stade d’angle fermable, la décision de traiter un patient asymptomatique présentant des angles étroits repose sur le jugement clinique de l’ophtalmologiste et sur l’évaluation précise de l’angle iridocornéen. Une profondeur de chambre antérieure inférieure à 2 mm, un contact iridotrabéculaire sur deux quadrants ou plus, voire des antécédents familiaux évidents, feront envisager très sérieusement une iridectomie. En cas d’iris plateau, une iridoplastie peut s’avérer nécessaire si l’iridotomie n’a pas permis l’approfondissement de l’angle.
Au stade de fermeture primitive de l’angle, une iridotomie (± iridoplastie) associée à un traitement médical hypotenseur permet de contrôler la maladie chez la plupart des patients.
Dans le glaucome primitif par fermeture de l’angle :
une fois l’iridotomie pratiquée, de nombreux patients nécessiteront un traitement complémentaire pour abaisser la PIO. Le traitement médical hypotenseur arrive souvent à contrôler la maladie ;
dans les cas réfractaires, souvent conséquences de SAP étendues sur plus de 180°, ou quand la neuropathie est évoluée, la chirurgie filtrante reste la méthode de référence pour abaisser la PIO à un niveau souhaitable.
La chirurgie du cristallin doit être sérieusement considérée dans la prise en charge de ces patients : chez tout patient ayant un angle fermé, s’il existe une cataracte, celle-ci doit être opérée, ce qui permet une réouverture de l’angle. En fonction du stade de la maladie, elle sera proposée seule ou combinée à une goniosynéchiolyse, voire à une trabéculectomie. En l’absence de cataracte, l’extraction du cristallin clair reste ouverte au débat. Une bonne stratégie pourrait être de pratiquer la phaco-exérèse et de réévaluer ensuite le contrôle pressionnel et l’état de l’angle : en cas d’HTO résiduelle, le traitement à envisager serait, selon les cas, médical ou chirurgical.
Retenir
Le GCFPA est une affection grave dont la fréquence est sous-estimée.
Son diagnostic repose sur la constatation de SAP à l’examen gonioscopique.
Il est indispensable de pratiquer un examen gonioscopique au moindre doute chez tout patient glaucomateux ou suspect de l’être.
Le traitement est avant tout chirurgical et peut, selon les cas, faire appel à l’iridotomie périphérique, à l’iridoplastie, à l’extraction du cristallin et à la chirurgie filtrante.
[1] Aptel F, Denis PH. Optical coherence tomography quantitative analysis of iris volume changes after pharmacologic mydriasis. Ophthalmology. 2010 ; 117 : 3-10.
[2] Boland MV, Zhang L, Broman AT, et al. Comparison of optic nerve head topography and visual field in eyes with open-angle and angle-closure glaucoma. Ophthalmology. 2008 ; 115 : 239-45.
[3] Congdon NG, Qi Y, Quigley HA, et al. Biometry and primary angle-closure glaucoma among Chinese, White, and Black populations. Ophthalmology. 1997 ; 104 : 1489-95.
[4] Foster PJ, Devereux JG, Alsbirk PH, et al. Detection of gonioscopically occludable angles and primary angle closure glaucoma by estimation of limbal chamber depth in Asians : modified grading scheme. Br J Ophthalmol. 2000 ; 84 : 186-92.
[5] Foster PJ, Buhrmann R, Quigley HA, Johnson GJ. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol. 2002 ; 86 : 238-42.
[6] Gazzard G, Friedman DS, Devereux JG, et al. A prospective ultrasound biomicroscopy evaluation of changes in anterior segment morphology after laser iridotomy in Asian eyes. Ophthalmology. 2003 ; 110 : 630-8.
[7] Gonzalez E, Peng PH, Lee R, et al. Anterior chamber depth, iridocorneal angle width, and intraocular pressure changes after phacoemulsification : narrow vs open iridocorneal angles. Arch Ophthalmol. 2011 ; 129 : 1283-90.
[8] Hamanaka T, Kasahara K, Takemura T. Histopathology of the trabecular meshwork and Schlemm’s canal in primary angle-closure glaucoma. Invest Ophthalmol Vis Sci. 2011 ; 52 : 8849-61.
[9] Hayashi K, Hayashi H, Nakao F, Hayashi F. Changes in anterior chamber angle width and depth after intraocular lens implantation in eyes with glaucoma. Ophthalmology. 2000 ; 107 : 698-703.
[10] He M, Friedman DS, Ge J, et al. Laser peripheral iridotomy in eyes with narrow drainage angles : ultrasound biomicroscopy outcomes (The Liwan Eye Study). Ophthalmology. 2007 ; 114 : 1513-9.
[11] How AC, Baskaran M, Kumar RS, et al. Changes in anterior segment morphology after laser peripheral iridotomy : an anterior segment optical coherence tomography study. Ophthalmology. 2012 ; 119 : 1383-7.
[12] Jacobi PC, Dietlein TS, Lüke C, et al. Primary phacoemulsification and intraocular lens implantation for acute angle-closure glaucoma. Ophthalmology. 2002 ; 109 : 1597-603.
[13] Kumar RS, Baskaran M, Chew PTK, et al. Prevalence of plateau iris in primary angle-closure suspects (an ultrasound biomicroscopy study). Ophthalmology. 2008 ; 115 : 430-4.
[14] Lai JS, Tham CC, Chan JS. The clinical outcomes of cataract extraction by phacoemulsification in eyes with primary angle-closure glaucoma (PACG) and co-existing cataract : a prospective case series. J Glaucoma. 2006 ; 15 : 47-52.
[15] Lteif Y, Berete-Coulibaly R, Labbé A, Lachkar Y. Résultats à moyen terme de la phacotrabéculectomie en deux sites avec microtrabéculectomie ajustable. J Fr Ophtalmol. 2008 ; 31 : 397-404.
[16] Muallem MS, Nelson GA, Osmanovic S, et al. Predicted refraction versus refraction outcome in cataract surgery after trabeculectomy. J Glaucoma. 2009 ; 18 : 284-7.
[17] Pavlin CJ, Ritch R, Foster FS. Ultrasound biomicroscopy in plateau iris syndrome. Am J Ophthalmol. 1992 ; 113 : 390-5.
[18] Pereira FA, Cronemberger S. Ultrasound biomicroscopic study of anterior segment changes after phacoemulsification and foldable intraocular lens implantation. Ophthalmology. 2003 ; 110 : 1799-806.
[19] Quek DT, Koh VT, Tan GS, et al. Blindness and long-term progression of visual field defects in Chinese patients with primary angle-closure glaucoma. Am J Ophthalmol. 2011 ; 152 : 463-9.
[20] Quigley HA. The iris is a sponge : a cause of angle closure. Ophthalmology. 2010 ; 117 : 1-2.
[21] Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006 ; 90 : 262-7.
[22] Quigley HA, Silver DM, Friedman DS, et al. Iris cross-sectional area decreases with pupil dilation and its dynamic behavior is a risk factor in angle closure. J Glaucoma. 2009 ; 18 : 173-9.
[23] Silver DM, Quigley HA. Aqueous flow through the iris-lens channel : estimates of differential pressure between the anterior and posterior chambers. J Glaucoma. 2004 ; 13 : 100-7.
[24] Tham CC, Kwong YY, Leung DY, et al. Phacoemulsification versus combined phacotrabeculectomy in medically controlled chronic angle closure glaucoma with cataract. Ophthalmology. 2008 ; 115 : 2167-73.
[25] Tham CC, Kwong YY, Leung DY, et al. Phacoemulsification versus combined phacotrabeculectomy in medically uncontrolled chronic angle closure glaucoma with cataracts. Ophthalmology. 2009 ; 116 : 725-3.
[26] Wang YX, Xu L, Yang H, Jonas JB. Prevalence of glaucoma in North China : the Beijing Eye Study. Am J Ophthalmol. 2010 ; 150 : 917-22.
C.-A. Ubaud, F. Matonti
Association d’un angle iridocornéen étroit, de synéchies antérieures périphériques et d’une neuropathie optique glaucomateuse provoquée par une pathologie oculaire sous-jacente non primitive responsable de la fermeture de l’angle.
Le diagnostic différentiel avec des glaucomes chroniques par fermeture primitive de l’angle, plus fréquents, peut présenter des difficultés, surtout dans les formes débutantes, frustes ou multifactorielles.
La classification et le diagnostic étiologique de ces glaucomes sont fondés sur l’identification des mécanismes provoquant la fermeture de l’angle : bloc pupillaire secondaire (sans prédispositions biométriques), attraction antérieure de la racine de l’iris (rubéose irienne, syndromes irido-cornéo-endothéliaux) ou poussée postérieure s’exerçant sur le plan iridocristallinien (kystes iridociliaires ou tumeurs, en particulier).
Le traitement comportera beaucoup moins systématiquement la réalisation d’une iridotomie que dans les glaucomes primitifs par fermeture de l’angle.
Différencier glaucome primitif à angle ouvert (GPAO) et glaucome chronique par fermeture secondaire de l’angle (GCFSA) est essentiel. Ce dernier est beaucoup plus rare, mais le confondre avec une autre forme de glaucome chronique aura pour conséquence un traitement inapproprié et donc la poursuite de l’évolution de la maladie.
La pathologie sous-jacente responsable peut non seulement engager le pronostic fonctionnel oculaire mais aussi, dans certains cas, le pronostic vital du patient.
La nomenclature actuellement admise [3] définit le glaucome chronique par fermeture de l’angle par l’association d’un angle iridocornéen étroit, de synéchies antérieures périphériques (SAP) et d’une atteinte glaucomateuse de la tête du nerf optique objectivée par une excavation et un déficit caractéristique du champ visuel.
Dans le GCFSA, la fermeture de l’angle résulte d’une pathologie oculaire sous-jacente. Ces GCFSA ont en commun avec le GPAO les éléments de la neuropathie optique glaucomateuse, mais s’en différencient en gonioscopie par une fermeture partielle ou totale de l’angle iridocornéen. Ils s’en distinguent également par l’existence de nombreuses causes sous-jacentes susceptibles de provoquer cette fermeture : iridopathies acquises (essentiellement rubéoses iriennes et syndromes irido-cornéo-endothéliaux), affections cristalliniennes et rétrocristalliniennes, ou diverses interventions thérapeutiques dirigées sur le segment postérieur.
Les GCFSA sont aussi à différencier des glaucomes chroniques par fermeture primitive de l’angle (GCFPA), bien plus fréquents : blocs pupillaires primitifs, configuration en iris plateau et, plus exceptionnellement, nanophtalmos. Les formes secondaires, à l’opposé des GCFPA, sont sans prédisposition biométrique, presque toujours unilatérales, sans facteur de risque ethnique (comme l’origine asiatique), et comportent toutes une origine pathologique sous-jacente.
La difficulté du diagnostic différentiel avec le GPAO concerne surtout certaines formes cliniques de GCFSA soit débutantes, soit d’étiologie multifactorielle, qui vont plus facilement se confondre avec un GPAO, et dont le diagnostic étiopathogénique risque d’être négligé. Dans ce cas, elles pourront simuler plus ou moins longtemps une évolution de GPAO par l’absence de symptômes fonctionnels, une élévation modérée de la pression intra-oculaire (PIO) au début, une atteinte progressive glaucomateuse de la tête du nerf optique et des déficits caractéristiques du champ visuel.
Dans d’autres cas, la symptomatologie sera plus bruyante, avec une PIO très élevée et une dégradation visuelle rapide, imposant sans tarder la reconnaissance de la cause.
Plus rarement, c’est la découverte de cette cause de la fermeture de l’angle qui amènera à poser le diagnostic de GCFSA.
Dans tous les cas, une démarche diagnostique étiopathogénique sera pratiquée afin de différencier un GCFSA d’un GPAO ou d’un autre type de glaucome. On adaptera les différents examens en fonction du contexte clinique et des éléments d’orientation. Ce bilan comprendra :
un bilan clinique du segment antérieur avec un examen à la lampe à fente de l’endothélium cornéen, du cristallin et de l’iris, une évaluation de la profondeur de la chambre antérieure par la recherche d’un signe de Van Herick et par différentes techniques de gonioscopie ;
un examen clinique complet du segment postérieur ;
enfin, le bilan paraclinique, en fonction des circonstances, peut inclure : un relevé du champ visuel ; une analyse en OCT des fibres nerveuses rétiniennes et du complexe maculaire cellulaire ganglionnaire ; une imagerie de l’endothélium cornéen, morphologique et avec comptage cellulaire ; une imagerie de l’angle iridocornéen (OCT du segment antérieur, biomicroscopie ultrasonore) ; une étude des paramètres biométriques de l’œil ; une échographie en mode B ; une angiofluorographie de l’iris ou du segment postérieur.
La classification des formes cliniques étiologiques la plus communément retenue actuellement est celle du tableau 14-4. Elle est fondée sur les mécanismes à l’origine de la fermeture secondaire de l’angle iridocornéen [23, 27]. Selon les cas, dans ces glaucomes, la fermeture de l’angle est provoquée par :
un bloc pupillaire s’exerçant au niveau du diaphragme iridocristallinien, avec pour causes les plus fréquentes les glaucomes phacomorphiques, les sub-luxations du cristallin du syndrome exfoliatif et des traumatismes oculaires ;
une poussée postérieure s’exerçant en arrière du plan iridocristallinien, dont les causes, plus rares, sont en particulier les kystes iridociliaires et les tumeurs oculaires ;
une attraction antérieure de la racine de l’iris secondaire liée essentiellement à une pathologie oculaire ischémique provoquant un glaucome néovasculaire (GNV), ou à un syndrome irido-cornéo-endothélial (ICE).
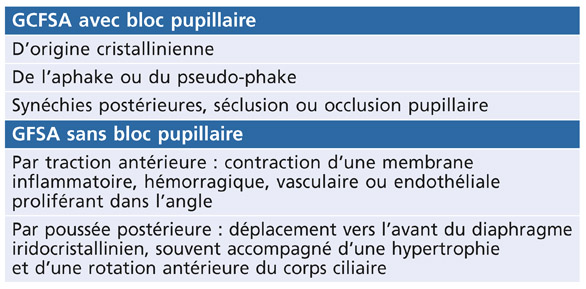
Tableau 14.4 – Classification des GCFSA.
Le mécanisme en est multifactoriel, mais essentiellement lié à une intumescence cristallinienne, dans un globe n’ayant pas de particularité biométrique prédisposant à une fermeture de l’angle. Le bloc pupillaire est présent et important. La distinction avec un GCFPA n’est pas toujours évidente. Cependant, une étude chinoise [12] montre un risque 4,3 fois supérieur de glaucome phacomorphique sur des yeux asiatiques dont la longueur axiale est inférieure à la moyenne (23,7 ± 1,5 mm dans la population chinoise prise comme référence) ; la moyenne de la longueur axiale des GCFSA est de 23,1 ± 0,9 mm. Une longueur axiale plus courte que la moyenne pourrait constituer ainsi un facteur prédisposant au risque de glaucome phacomorphique.
Iridotomie périphérique puis phaco-émulsification, ou phaco-émulsification d’emblée [13], traitements dont l’ordre est à discuter en fonction de la profondeur de l’angle et de la chambre antérieure.
Une sub-luxation du cristallin est exceptionnellement à l’origine d’un bloc pupillaire. Elle est le plus souvent minime, sans conséquence délétère à court terme et compatible avec une surveillance clinique régulière attentive.
Dans certains cas, associée à une hernie vitréenne transpupillaire, elle peut être à l’origine d’un mécanisme de blocage pupillaire qui, progressivement, risque d’entraîner une hypertension oculaire et donc un glaucome chronique.
Forme clinique fréquente, voire majoritaire dans certains pays, le syndrome exfoliatif est à l’origine de glaucomes à angle ouvert comme de glaucomes par fermeture de l’angle. Sur les 60 à 70 millions de personnes dans le monde ayant ce syndrome, 5 à 6 millions auraient un glaucome. La plupart d’entre eux ne sont ni diagnostiqués, ni suspectés [17].
L’atteinte de tous les tissus du segment antérieur de l’œil prédispose à diverses complications incluant glaucome à angle ouvert, glaucome par fermeture primitive de l’angle, phacodonésis et subluxation du cristallin. Les altérations zonulaires, autant au niveau de leur insertion capsulaire que de leur ancrage dans l’épithélium ciliaire non pigmenté, aboutissent à un phacodonésis ou à un déplacement vers le bas du cristallin. Particulièrement en décubitus ventral, un déplacement vers l’avant du cristallin peut produire un bloc pupillaire ou même un bloc ciliaire prédisposant au glaucome par fermeture de l’angle, fréquent donc dans les yeux présentant un syndrome exfoliatif [18, 22].
Les déplacements post-traumatiques du cristallin font partie des causes les plus fréquentes de glaucome par fermeture de l’angle après un traumatisme oculaire non perforant [21].
Microsphérophakie congénitale [24], souvent familiale, isolée [11] ou intégrée dans un syndrome de Weill-Marchesani ou de Marfan [6].
Homocystinurie.
Iridectomie périphérique, parfois deux à 180° l’une de l’autre en cas de risque d’obstruction de l’iridotomie périphérique initiale par le cristallin subluxé, et/ou chirurgie du cristallin en particulier dans le syndrome exfoliatif. Une cycloplégie peut être discutée pour lever le bloc pupillaire des microsphérophakies en tendant la zonule. Les myotiques sont à considérer avec prudence car ils peuvent aggraver le tableau clinique : augmentation du bloc pupillaire, bascule du corps ciliaire en avant, relâchement de la zonule.
Un bloc pupillaire peut se créer :
chez l’aphake, par blocage pupillaire hyaloïdovitréen et exceptionnellement par l’obstruction de l’iridectomie, ou après capsulotomie au laser YAG ;
sur un implant de chambre antérieure, en l’absence d’iridectomie, avec une chambre antérieure profonde au centre et un angle fermé (aspect de pneu irien périphérique).
Lever le bloc pupillaire par une iridotomie périphérique au laser.
Lors d’une inflammation oculaire, la rupture de la barrière hémato-aqueuse avec présence de fibrine et augmentation des protéines dans l’humeur aqueuse peut entraîner la formation de synéchies postérieures, une séclusion pupillaire, un iris bombé (« iris tomate ») et donc une fermeture de l’angle.
Le GNV survient essentiellement dans un contexte ischémique rétinien [5]. Toutes les rétinopathies ischémiques, en particulier la rétinopathie diabétique ou les formes ischémique d’occlusion de la veine centrale de la rétine (OVCR), peuvent se compliquer d’une ischémie rétinienne et du segment antérieur, à l’origine de néovaisseaux iriens angulaires.
Le diagnostic précoce de GNV est souvent difficile en cas de rubeosis insipiens [7], de GPAO préexistant, de néovaisseaux débutant dans l’angle (10 % des cas de GNV dans les OVCR) et d’ischémie rétinienne non affirmée (fig. 14-21).
Devant une ischémie rétinienne périphérique sans cause oculaire décelable, il est impératif d’envisager le diagnostic d’occlusion carotidienne, souvent bilatérale et asymétrique. Elle sera recherchée systématiquement par un écho-Doppler des troncs supra-aortiques.
Le plus précocement possible, traitement des zones ischémiques par photocoagulation rétinienne, associée à une injection intravitréenne d’agents anti-VEGF et à un traitement hypotonisant oculaire. Une chirurgie filtrante, la pose d’un système de drainage (drain) ou une cyclodestruction, si nécessaire, pourraient être ainsi réalisées dans de meilleures conditions [8].
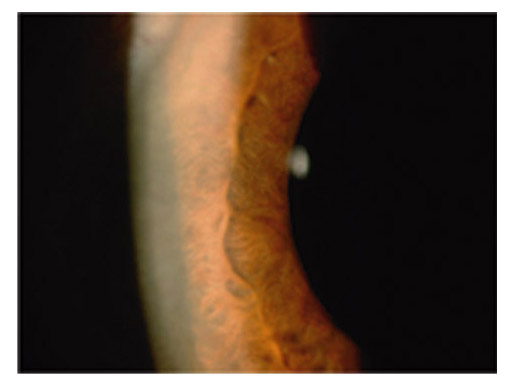
Fig. 14-21 GNV débutant : néovaisseaux iriens à peine décelables au bord pupillaire.
Ce syndrome regroupe trois variantes cliniques d’une affection caractérisée par une perte de contrôle du cycle cellulaire endothélial cornéen [19] et une migration de ces cellules au-delà de la ligne de Schwalbe, vers la base du corps ciliaire, le trabéculum et l’iris : le syndrome de Chandler, l’atrophie essentielle de l’iris, le syndrome du nævus irien de Cogan-Reese.
Ils présentent un certain nombre de points communs :
unilatéralité quasi constante ;
début entre 20 et 50 ans de façon unilatérale, plus souvent chez une femme ;
très rarement familial ;
endothélium cornéen anormal, dont les cellules migrent vers le trabéculum en arrière de l’anneau de Schwalbe ;
aspect endothélial en « argent battu », souvent initialement discret ;
classiquement dans l’angle, larges SAP en avant de l’anneau de Schwalbe ;
évolution vers une atrophie de l’iris et un œdème cornéen ;
GCFSA chez 50 % des patients ayant un syndrome ICE.
L’importance respective de l’atrophie irienne et des modifications cornéennes différencient les trois entités classiques :
le syndrome de Chandler (fig. 14-22) réalise 50 % des cas de syndrome ICE. L’atrophie de l’iris y est minime, associée à une corectopie, les atteintes cornéennes et angulaires étant prédominantes ;
dans l’atrophie essentielle de l’iris (fig. 14-23), l’atrophie sévère et progressive du stroma et de l’épithélium pigmentaire irien provoque hétérochromie, corectopie, ectropion uvéal et trous iriens ;
dans le syndrome de Cogan-Reese (fig. 14-24), l’atrophie de l’iris est plus modérée. La présence de nodules bruns pédonculés ou de lésions pigmentées diffuses à la surface antérieure de l’iris caractérise cette variante.
Le glaucome est en général plus sévère dans l’atrophie essentielle de l’iris et le syndrome de Cogan-Reese. Ces pathologies ICE peuvent imiter un GPAO lorsque l’atteinte irienne et cornéenne est très discrète. La confirmation peut en être apportée par la microscopie spéculaire et la microscopie confocale in vivo en cas de doute diagnostique [25].
Ces syndromes ICE sont à distinguer de l’exceptionnel iridoschisis et du syndrome d’Axenfeld congénital et bilatéral.
Retenir l’inefficacité des myotiques et des trabéculoplasties au laser et l’incertitude sur l’effet des analogues de prostaglandines. Le recours à la chirurgie filtrante perforante, voire aux valves et/ou aux cyclo-affaiblissements, est souvent nécessaire.

Fig. 14-22 a. Syndrome de Chandler débutant : légère déformation pupillaire inférieure de l’œil gauche (les figures a,b,c concernent le même patient). b. Microscopie spéculaire : œil droit sain. c. Microscopie spéculaire : œil gauche atteint. d. SAP inférieure en UBM œil gauche.

Fig. 14-23 a. Atrophie essentielle de l’iris à un stade débutant. b. Gonioscopie : SAP à 12 heures chez la même patiente.
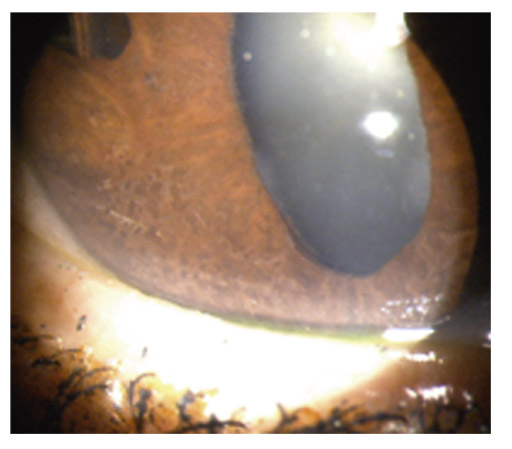
Fig. 14-24 Syndrome de Cogan-Reese. (Cliché : E. Sellem.)
Les mécanismes sont volontiers multifactoriels, avec la constitution progressive de SAP, souvent associée à des synéchies postérieures (séclusion pupillaire) et à une effusion uvéale ou une congestion ciliaire. À la différence des SAP des GCFPA, elles se situent ici plus fréquemment au niveau de l’angle inférieur et sont inhomogènes en forme et hauteur.
SAP secondaires après crise aiguë de glaucome par fermeture de l’angle résolutive.
Invasion épithéliale [28].
Complications de la trabéculoplastie au laser à l’argon.
Aniridies congénitales [11].
Dystrophie endothéliale polymorphe postérieure.
Traumatismes oculaires [21], après hyphéma et inflammation, et dont le diagnostic est souvent tardif.
Les kystes iridociliaires de l’épithélium pigmentaire sont les plus fréquents et le plus souvent périphériques (73 %) au niveau du sulcus iridociliaire. Ils se développent plutôt chez des patients jeunes, plus souvent de sexe féminin, de localisation multiple dans environ 40 % des cas, et n’entraînent que rarement des complications en dehors du GCFSA (glaucome par fermeture de l’angle dans 2 % des cas) [2, 14, 26]. L’examen UBM [27] permet le diagnostic différentiel avec un GPAO, un iris plateau et une tumeur. Le traitement par cystostomie transpupillaire ou iridocystostomie au laser Nd-YAG a été proposé [9], ou par iridorétraction au laser à l’argon.
Au niveau du segment postérieur, les mélanomes choroïdiens primitifs, ainsi que les rétinoblastomes et les métastases oculaires, sont les plus fréquents : au déplacement vers l’avant du diaphragme iridocristallinien par poussée postérieure peuvent s’associer d’autres mécanismes, inflammatoires et néovasculaires, entraînant la fermeture secondaire de l’angle [20].
Cette forme rare et sévère de glaucome survient habituellement sous une forme aiguë après une chirurgie oculaire (2 à 4 % des trabéculectomies), le plus souvent chez des patients aux antécédents de fermeture de l’angle ou porteurs de SAP. Dans ces circonstances, le diagnostic différentiel se pose donc peu. Son mécanisme est controversé : fausse route de l’humeur aqueuse déviée postérieurement et bloc ciliaire [15], ou expansion choroïdienne et conductivité réduite du vitré [16] dans un œil de petite taille.
Le traitement médical associe une cycloplégie puissante et des agents hyperosmotiques. Les myotiques sont contre-indiqués. Une hyaloïdotomie antérieure au laser Nd-YAG doit être pratiquée si l’œil est aphake ou pseudo-phake. La réalisation d’un cyclo-affaiblissement est parfois efficace. Enfin, en cas d’échec, une procédure chirurgicale s’impose, associant une vitrectomie, une hyaloïdotomie et une technique d’approfondissement de la chambre antérieure.
En dehors des tumeurs intra-oculaires déjà citées, de nombreuses causes peuvent provoquer des effusions uvéales ou des décollements de rétine secondaires : cyclites et uvéites postérieures chroniques, augmentation de la pression veineuse choroïdienne (nanophtalmos, OVCR, communication artérioveineuse), maladie de Coats [4].
Dans ces deux affections, la contraction du tissu rétrolental peut entraîner une fermeture progressive de l’angle iridocornéen et un glaucome par fermeture de l’angle. Bien que ces glaucomes puissent survenir un peu tardivement dans l’enfance (2 % des rétinopathies des prématurés sévères sont suivies d’un glaucome dans les six premières années de la vie), le contexte clinique local et la notion de prématurité rendent le diagnostic différentiel avec un glaucome juvénile relativement aisé [1].
Le contexte clinique laisse peu de place à une confusion diagnostique avec un GPAO. La question peut éventuellement se poser en cas de photocoagulation panrétinienne augmentant le volume choriorétinien et limitant le drainage veineux. De même, après une chirurgie rétinienne, une fermeture chronique de l’angle peut s’expliquer par l’inflammation entraînant une bascule vers l’avant du corps ciliaire, et/ou par l’effet mécanique de l’indentation sur la position du cristallin et sur le drainage veineux choroïdien. La présence de gaz ou d’huile de silicone laisse, en revanche, peu de place au doute diagnostique.
Cette situation, actuellement exceptionnelle, le plus souvent liée à une fuite par l’incision, doit être résolue rapidement. Dans le cas contraire pourront se développer des SAP et un GCFSA à différencier ultérieurement d’un glaucome à angle ouvert du pseudo-phake.
Retenir
Multiplicité des étiologies ouvrant sur toutes les pathologies oculaires.
Multiplicité des mécanismes possibles avec ou sans bloc pupillaire.
Évoquer en particulier le glaucome phacomorphique, le syndrome exfoliatif, le GNV et les syndromes ICE.
Atteinte unilatérale habituelle.
Importance de l’examen comparatif de l’œil controlatéral.
Diagnostic différentiel avec GPAO : attention aux formes débutantes et frustes de GCFSA.
Importance de la gonioscopie et de l’imagerie du segment antérieur.
Contexte général du patient souvent impliqué (âge, sexe, diabète, HTA).
Importance vitale des néoplasies oculaires.
[1] Bremen DL, Rogers DL, Good WV, et al. Glaucoma in the Early Treatment for Retinopathy of Prematurity (ETROP) study. J AAPOS. 2012 ; 16 : 449-52.
[2] Fine N, Pavlin CJ. Primary cysts in the iridociliary sulcus. Can J Ophthalmol. 1999 ; 34 : 325-9.
[3] Foster PJ, Buhrmann R, Quigley HA. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol. 2001 ; 84 : 186-92.
[4] Ghorbanian S, Jaulim A, Chatziralli IP. Diagnosis and treatment of Coat’s disease : a review of the literature. Ophthalmologica. 2012 ; 227 : 175-82.
[5] Hayreh SS. Neovascular glaucoma. Prog Retin Eye Res. 2007 ; 26 : 470-85.
[6] Hoffjan S. Genetic dissection of Marfan syndrome and related connective tissue disorders : an update 2012. Mol Syndromol. 2012 ; 3 : 47-58.
[7] Inoue M, Azumi A, Shirabe H, Yamamoto M. Iridopathy in eyes with proliferative diabetic retinopathy ; detection of early stage of rubeosis iridis. Ophthalmologica. 1998 ; 212 : 15-8.
[8] Kodjikian L. Neovascular glaucoma treatement in 2012 : role of anti-VEGF agents. J Fr Ophtalmol. 2013 ; 5 : 461-5.
[9] Kuchenbecker J. Laser iridocystostomy for bilateral acute angle-closure glaucoma secondary to iris cysts. Am J ophthalmol. 2000 ; 129 : 391-3.
[10] Kumar A, Duvvari MR, Prabhakaran VC, et al. A homozygous mutation in LTBP2 caused isolated microspherophakia. Hum Genet. 2010 ; 128 : 365-71.
[11] Lee H, Khan R, O’Keefe M. Aniridia : current pathology and management. Acta Ophthalmol. 2008 ; 86 : 708-15.
[12] Lee JW, Lam RF, Wong BK, et al. Retrospective analysis of the risk factors for developing phacomorphic glaucoma Indian. J Ophthalmol. 2011 ; 59 : 471-4.
[13] Lee SJ, Lee CK, Kim WS. Long-term therapeutic efficacy of phacoemulsification with intraocular lens implantation in patients with phacomorphic glaucoma. J Cataract Refract Surg. 2010 ; 36 : 783-9.
[14] Lois N, Shields CL, Shields JA, Mercado G. Primary cysts of the iris pigment epithelium. Clinical features and natural course in 234 patients. Ophthalmology. 1998 ; 105 : 1879-85.
[15] Lundy DC. Ciliary block glaucoma. Focal points : clinical modules for ophthalmologists. San Francisco, American Academy of Ophthalmology, 1999, module 3.
[16] Quigley HA, Friedman DS, Congdon NG. Possible mecanisms of primary angle-closure and malignant glaucoma. J Glaucoma. 2003 ; 12 : 167-80.
[17] Ritch R. From exfoliation syndrome to exfoliative glaucoma. In : Hollo G, Konstas AGP (eds). Exfoliation syndrome and exfoliative glaucoma. Dogma, 2008 : 19-23.
[18] Ritch R. Why is glaucoma associated with exfoliation syndrome. Prog Retin Eye Res. 2003 ; 22 : 253-75.
[19] Robert AM, Renard G, Robert L, Bourges JL. The irido-corneo-endothelial syndrome. The loss of the control of corneal endothelial cell cycle. A review. Pathol Biol (Paris). 2013 ; 61 : 75-82.
[20] Rohrbach JM, Grüb M, Schlote T. Neoplasic secondary glaucomas. Klin Monbl Augenheilkd. 2005 ; 222 : 788-96.
[21] Schlote T, Rohrbach M. Traumatic glaucoma. A Survey. Klin Monbl Augenheilkd. 2005 ; 222 : 772-82.
[22] Schlötzer-Schrehardt U, Naumann GOH. Morphology of exfoliation syndrome. In exfoliative syndrome and exfoliative glaucoma. In : Hollo G, Konstas AGP (eds). Exfoliation syndrome and exfoliative glaucoma. Dogma, 2008 : 35-43
[23] Sellem E. Les mécanismes des glaucomes par fermeture de l’angle. J Fr Ophtalmol. 2004 ; 27 :693-6.
[24] Senthil S, Rao HL, Hoang NT, et al. Glaucoma in microspherophakia : presenting features and treatment outcomes. J Glaucoma. 2012 ; 10 [Epub ahead of print].
[25] Sheppard JD, Lattanzio FA, Williams PB, et al. Confocal microscopy used as the definite, early diagnostic method in Chandler syndrome. Cornea. 2005 ; 24 : 227-9.
[26] Shields JA. Primary cysts of the iris. Trans Am Ophthalmol Soc. 1981 ; 79 : 771-809.
[27] Tello C, Rothman R, Ishikawa H, Ritch R. Differential diagnosis of the angle closure glaucoma. Ophthalmol Clin North Am. 2000 ; 13 : 443-53.
[28] Zargorski Z, Shrestha HG, Lang GK, Nauman GO. Secondary glaucoma caused by intraocular epithelial invasion. A clinico-pathologic study of 30 patients. Klin Monbl Augenheilkd. 1988 ; 193 : 16-20.
C. Vignal-Clermont
Devant une excavation papillaire pathologique et une pression intra-oculaire (PIO) normale, le principal risque est de méconnaître une neuropathie optique non glaucomateuse, en particulier compressive, et de retarder son traitement.
L’existence d’une excavation du disque optique ne suffit pas pour affirmer le diagnostic de neuropathie optique glaucomateuse. Le terrain, l’histoire et l’examen cliniques, incluant la mesure de la PIO, la gonioscopie, la pachymétrie et l’aspect du champ visuel, sont des éléments déterminants.
La constatation d’une discordance entre la structure du disque optique (aspect du fond d’œil et OCT) et la fonction visuelle (acuité et champ visuels) doit faire suspecter une neuropathie optique non glaucomateuse et demander une imagerie des voies visuelles.
En cas de suspicion de neuropathie optique non glaucomateuse, un examen d’imagerie des voies visuelles permet d’éliminer une compression et de préciser le calibre et le signal des nerfs optiques et du chiasma. En l’absence de contre-indication, il faut privilégier l’IRM cérébrale et orbitaire avec des coupes dans plusieurs plans de l’espace (sagittal, axial et coronal), sans et avec injection de gadolinium, et en supprimant le signal de la graisse orbitaire.
Les neuropathies optiques (NO) ont comme conséquence commune une diminution des axones des cellules ganglionnaires qui s’installe après quelques semaines à quelques mois d’évolution [15]. Cette perte en fibres visuelles concerne toutes les atteintes du nerf optique quelle que soit leur étiologie ; elle s’accompagne d’une modification de l’aspect de la papille avec, à un degré variable selon le mécanisme, l’apparition d’une pâleur et une augmentation de l’excavation du disque optique (DO). L’excavation du disque optique n’est donc pas l’apanage de la neuropathie optique glaucomateuse [3].
Les difficultés diagnostiques sont doubles :
le glaucome étant la neuropathie optique la plus fréquente, le premier risque est de porter par excès le diagnostic de glaucome à pression normale (GPN) devant une papille pâle et excavée, lorsque la PIO et la pachymétrie sont normales ;
lors du suivi d’un patient glaucomateux connu et traité, équilibré sur le plan pressionnel, si l’évolution de la fonction visuelle (acuité et/ou champ visuels) ou de l’aspect de la papille est discordante par rapport aux valeurs de la PIO, le diagnostic de NO non glaucomateuse intriquée doit être suspecté.
Les difficultés diagnostiques sur le seul aspect de la papille ont été soulignées dans plusieurs publications [9-11, 23, 28-30] et tous les auteurs insistent sur l’importance de l’interrogatoire et de l’examen clinique avec examen soigneux de la papille et de l’angle iridocornéen, incluant le champ visuel. L’OCT peut également apporter des arguments diagnostiques très utiles. Parfois, une imagerie cérébrale et orbitaire sera nécessaire pour préciser le calibre et le signal des nerfs optiques et du chiasma, et surtout rechercher un élément compressif.
L’étude la plus récente rapportant les difficultés diagnostiques devant une papille pâle et excavée a été publiée en 2011 [23]. Trente-trois cliniciens, tous experts en glaucome et neuro-ophtalmologie, ont évalué sans autre élément clinique les stéréophotographies d’un des DO de 60 patients (15 DO normaux, 15 glaucomes, 15 atrophies optiques dominantes et 15 neuropathies optiques de Leber). Le diagnostic était correct pour 85 % des glaucomes mais seulement 27 % des atrophies optiques dominantes (AOD) et 16 % des neuropathies de Leber. Le diagnostic de glaucome a été porté par excès dans 39 % des cas, dont 19 % concernaient des AOD, 15 % des Leber et 5 % des papilles normales. Les AOD ont été diagnostiquées comme un glaucome dans 48 % des cas et les NO de Leber dans 39 % des cas. La conclusion des auteurs est que l’aspect du nerf optique ne suffit pas et que l’histoire et l’examen cliniques sont des éléments indispensables à un diagnostic correct. En 1998 et 1999, Greenfield et son équipe ont comparé rétrospectivement 52 yeux de 29 patients avec un GPN et 44 yeux de 28 patients avec des lésions compressives [9, 10]. La comparaison portait non seulement sur l’aspect du fond d’œil mais aussi sur le terrain et les données cliniques, incluant l’acuité et le champ visuels. Les patients avec un GPN sont significativement plus âgés, ont une meilleure acuité visuelle, leur excavation papillaire est plus verticale, avec des hémorragies péripapillaires et un anneau neurorétinien moins pâle. Les déficits fasciculaires des glaucomateux sont alignés sur le méridien horizontal (cet élément est spécifique du glaucome à 84 % dans cette étude). Les auteurs concluent qu’une acuité inférieure à 5/10, un déficit campimétrique respectant le méridien vertical, un DO plus pâle qu’excavé et un âge inférieur à 50 ans sont spécifiques à 77, 81, 90 et 93 % respectivement d’une NO compressive non glaucomateuse. Aucune lésion compressive n’a été retrouvée chez les patients présentant un GPN, soulignant l’importance de l’histoire clinique et du champ visuel en complément de l’examen des papilles, afin d’éviter des examens neuroradiologiques inutiles. Deux autres publications dues à Trobe et son équipe font état des mêmes difficultés ; dans la première [29], trois ophtalmologistes ont examiné, sans connaître l’histoire clinique et le champ visuel, les photographies des papilles de patients glaucomateux et de porteurs de NO non glaucomateuses ; 44 % des NO non glaucomateuses ont été étiquetées glaucome par au moins un des observateurs Au niveau du fond d’œil, les deux éléments les plus utiles pour faire la distinction entre ces entités sont la pâleur diffuse de l’anneau neurorétinien (ANR) (spécifique de la NO non glaucomateuse à 94 %) et l’absence focale ou diffuse de l’ANR (spécifique du glaucome à 87 %). La même équipe a publié la même année une étude incluant cinq observateurs et 163 photos non stéréoscopiques de fond d’œil sans que l’histoire clinique soit disponible [30]. Si les neuropathies glaucomateuses sont correctement diagnostiquées dans plus de 80 % des cas, les neuropathies optiques ischémiques séquellaires et les atteintes héréditaires et compressives sont diagnostiquées comme glaucome dans un cas sur deux.
En 2011, Gupta et al. [11] se sont intéressés à l’apport de l’OCT dans le diagnostic étiologique des neuropathies optiques avec excavation papillaire, en comparant les patients présentant un glaucome et une neuropathie optique d’une autre origine. Onze patients avec excavation du nerf optique non glaucomateuse [six névrites optiques, une anomalie congénitale, trois séquelles de neuropathie optique ischémique antérieure (NOIA) non artéritique et une neuropathie optique par hypovolémie (shock-induced)] et 12 patients avec une excavation papillaire glaucomateuse (8 GPAO et 4 GPN) ont été étudiés prospectivement entre 2007 et 2008 avec l’OCT Stratus® et le protocole fast RNFL. Les patients non glaucomateux sont plus jeunes (43 ± 19,8 ans versus 67,8 ± 12,5 ans, p < 0,05) et, pour une valeur identique du rapport C/D mesuré à l’OCT, lorsque l’on compare des patients appartenant à chaque groupe, l’épaisseur des fibres visuelles dans le quadrant nasal et le volume maculaire sont plus faibles chez les patients non glaucomateux. Pour une valeur identique d’épaisseur moyenne de la couche des FNR, les mesures des FNR en nasal et temporal sont plus faibles dans les NO non glaucomateuses. Ces données confirment que dans le glaucome, il existe une perte en fibres touchant le plus souvent les quadrants supérieur et inférieur avec une excavation verticale et une disparition de la règle ISNT alors que dans les NO non glaucomateuses, la topographie de la perte en fibres dépend du mécanisme et de l’étiologie de la neuropathie [22].
Les données de la littérature soulignent les éléments qui permettent de différencier une neuropathie optique non glaucomateuse d’un glaucome et qui doivent être appréciés lors du bilan initial de chaque neuropathie. Ces arguments sont résumés dans le tableau 14-5.
En plus de l’examen clinique, l’interrogatoire doit préciser les circonstances et la cinétique de l’installation de la gêne visuelle, ainsi que le terrain (antécédents, âge du patient, facteurs de risque vasculaire et symptômes associés).
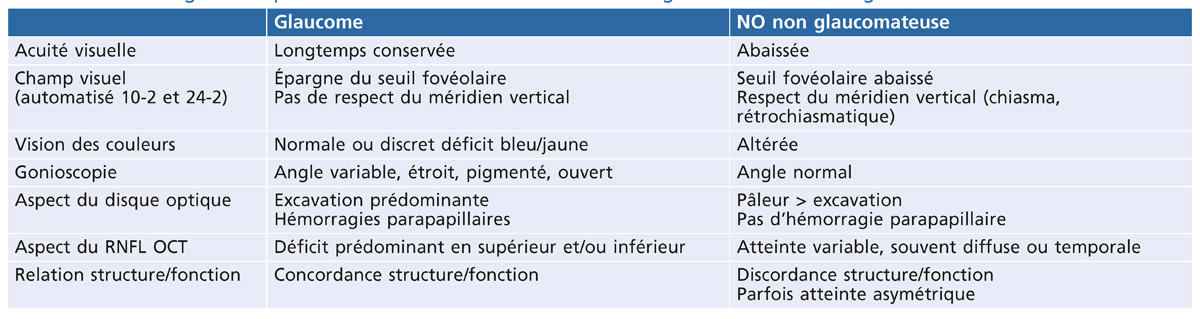
Tableau 14.5 – Arguments permettant de différencier une NO non glaucomateuse d’un glaucome.
Certaines entraînent une perte en fibres sectorielle avec un déficit fasciculaire du champ visuel dans le territoire correspondant ; le champ visuel de ces patients simule alors un déficit glaucomateux. L’examen des pupilles est en règle normal, sans déficit pupillaire afférent relatif du côté de l’atteinte. Le diagnostic est établi facilement par l’examen du fond d’œil après dilatation, mettant en évidence la papille non glaucomateuse et l’anomalie rétinienne ou chororétinienne responsable du déficit (eCas clinique 14-3).
Parfois, après un épisode de bas débit dans l’artère centrale, il existe une baisse visuelle avec une papille qui devient pâle et des artères un peu grêles. Le champ visuel montre alors souvent un déficit diffus ou un scotome central homolatéral. L’examen par OCT peut mettre en évidence une désorganisation de la rétine interne évoquant une ischémie rétinienne ; l’électrophysiologie (ERG et PEV) permettra de confirmer l’atteinte de la rétine dans les cas les plus difficiles.
Séquelle de nodule cotonneux avec occlusion artériolaire en temporal inférieur de la papille droite.

Fig. A Champ visuel seuil 24-2 : déficit arciforme nasal supérieur simulant une atteinte glaucomateuse.

Fig. B Cliché couleur : la papille droite est pâle en temporal avec quelques exsudats secs parapapillaires.

Fig. C Cliché en lumière bleu qui visualise le déficit en fibres visuelles temporal inférieur et coupe OCT montrant l’amincissement rétinien lié à la séquelle vasculaire.

Fig. D Mesure du RNFL qui confirme le déficit dans le cadrant temporal inférieur. (Collection du docteur Affortit.)
Les principales neuropathies optiques qui s’accompagnent d’une excavation papillaire (en dehors des excavations et des anomalies papillaires congénitales) sont énumérées dans le tableau 14-6 [20, 26].

Tableau 14.6 – Principales neuropathies optiques pouvant entraîner une excavation papillaire pathologique.
Parmi les anomalies congénitales de la papille, les colobomes, qui résultent d’un défaut de fermeture de la fente fœtale, entraînent une excavation au sein de la papille ; elle est le plus souvent située en inférieur avec un déficit campimétrique correspondant [21]. Dans les fossettes colobomateuses, il existe une excavation sectorielle du DO avec dans la moitié des cas un déficit du champ visuel paracentral ou arciforme qui peut poser des problèmes diagnostiques (fig. 14-25). La principale complication des fossettes est la survenue d’un décollement séreux rétinien qui complique la fossette dans 25 à 75 % des cas. Certaines dysversions papillaires peuvent s’accompagner d’une excavation. L’analyse du DO chez ces patients myopes et astigmates, avec un champ visuel souvent anormal (déficit bitemporal ne respectant pas le méridien vertical), est difficile et leur suivi doit être rigoureux afin de ne pas méconnaître un glaucome débutant [7]. Enfin, des cas d’hypoplasie du nerf optique avec une excavation papillaire anormale, parfois associés à des anomalies cérébrales de la ligne médiane (syndrome de Morsier) ou à une leucomalacie périventriculaire, ont été décrits [14].

Fig. 14-25 Fossette colobomateuse de la papille. (Cliché : E. Sellem.)
L’atrophie optique dominante ou maladie de Kjer et la neuropathie optique de Leber peuvent se traduire au niveau du fond d’œil par des papilles pâles et excavées et poser un réel problème diagnostique avec le glaucome.
C’est la plus fréquente des neuropathies optiques héréditaires avec une prévalence estimée à environ 1/10 000-1/50 000:individus. Elle est liée principalement à une mutation du gène OPA1, situé en 3q28-29, et impliquée dans la dynamique du réseau mitochondrial ; plus de 100:mutations différentes sont recensées. L’AOD a une pénétrance incomplète et une expressivité intra- et interfamiliale variable. Elle est habituellement diagnostiquée dans l’enfance, mais des formes congénitales et tardives (après 60 ans) sont aussi décrites. La baisse de l’acuité visuelle est progressive et s’accompagne d’une dyschromatopsie le plus souvent dans l’axe bleu-jaune, mais parfois dans l’axe rouge/vert. Le signe cardinal de la maladie est l’atrophie optique visible au fond d’œil. La pâleur papillaire est l’anomalie la plus fréquente ; elle est localisée en temporal dans un cas sur deux et globale dans l’autre moitié [31]. La présence d’une excavation profonde est décrite chez 21 % des patients avec un rapport C/D supérieur à 0,5 chez 50 % [2]. Cependant, la pente de l’excavation est classiquement douce et progressive avec la présence d’un croissant gris temporopapillaire et une atrophie péripapillaire fréquente. Outre l’histoire clinique, les éventuels antécédents familiaux, la pâleur papillaire et l’aspect de l’excavation, les autres éléments du diagnostic différentiel sont la présence d’une baisse d’acuité visuelle en rapport avec un scotome cæcocentral bilatéral et souvent symétrique ainsi que la perte en fibres localisée dans le secteur temporal et mise en évidence à l’OCT (eCas clinique 14-4).
Patiente de 50 ans se plaignant d’une baisse visuelle très progressive. Acuité à 5/10 Parinaud 2 faible OD et OG. Deux de ses cinq enfants ont une vision un peu basse. Le bilan génétique va mettre en évidence une mutation dans l’exon 16 du gène OPA1 et permettre de faire le diagnostic d’atrophie optique dominante.

Fig. A, B L’examen du fond d’œil montre des papilles pâles en temporal, assez bien colorées en nasal, avec un croissant atrophique péripapillaire plus important à droite. L’excavation papillaire est marquée mais en pente douce.

Fig. C, D Champ visuel seuil central 24-2 : abaissement du seuil fovéal à 29 dB à droite et 25 dB à gauche avec un déficit prédominant dans le quadrant temporal supérieur et ne respectant pas le méridien vertical.

Fig. E, F L’OCT retrouve une perte en fibres dans le quadrant temporal au niveau de chaque œil. Il existe par ailleurs une dyschromatopsie bleu/jaune d’axe tritan (F).
Elle pose moins de problèmes diagnostiques ; elle survient dans 70 % des cas chez un homme jeune, encore que des cas tardifs soient décrits. Elle se traduit par une baisse d’acuité visuelle sévère, unilatérale, indolore, rapidement progressive voire brutale. La bilatéralisation survient au bout de quelque mois avec une acuité effondrée et un scotome cæcocentral bilatéral. Au stade aigu, la papille est hyperhémiée, avec des télangiectasies péripapillaires, un aspect pseudo-œdémateux et une tortuosité des vaisseaux rétiniens. Au stade tardif, les papilles sont atrophiques et globalement pâles, avec une excavation en pente douce. La NO héréditaire de Leber est en rapport avec une mutation de l’ADN mitochondrial qui a comme conséquence une atteinte de l’activité du complexe 1 de la chaîne respiratoire avec altération de la production d’ATP par la mitochondrie. Trois mutations primaires (11778, 14484 et 3460) sont décrites et représentent 95 % des cas. Le diagnostic différentiel avec le GPN se pose peu : dans la moitié des cas, une hérédité mitochondriale avec une transmission maternelle de la maladie est retrouvée, les patients sont souvent jeunes, l’acuité visuelle est effondrée, l’ANR est atrophique et pâle en totalité [25]. Cependant, chez les patients plus âgés, lorsqu’il existe une hypertonie oculaire associée, le diagnostic peut être difficile et retardé (eCas clinique 14-5).
Monsieur V. est adressé pour avis sur une baisse visuelle droite isolée, indolore, rapidement progressive, ayant débuté trois mois auparavant. L’acuité visuelle est effondrée à droite à 1/10 faible et à 8/10 à gauche. La PIO est mesurée à 35 à droite et 25 à gauche à l’aplanation, l’angle est ouvert et la pachymétrie est mesurée à 556 à droite et 560 à gauche. Le patient est traité par collyre hypotonisant. Six semaines plus tard, la vision est inchangée à droite, réduite à 3/10 faible à gauche.

Fig. A, B Examen du fond d’œil : papille pâle en temporal et inférieur à droite avec une excavation en pente douce. À gauche, papille mieux colorée, avec une hyperhémie nasale et une discrète pâleur temporale.

Fig. C, D Le champ visuel montre un déficit cæcocentral absolu à droite, moins marqué à gauche où le déficit affleure le point de fixation. Le champ visuel et l’hyperhémie papillaire nasale sont discordants avec le diagnostic de glaucome. L’analyse de l’ADN mitochondrial retrouve une mutation 11778 homoplasmique et confirme le diagnostic de neuropathie de Leber.

Fig. E L’OCT RNFL de la papille droite montre l’évolution de l’épaisseur des fibres visuelles avec un amincissement démarrant en temporal.
Elle se traduit au stade aigu par une baisse visuelle brutale, souvent profonde, avec au niveau du fond d’œil un œdème papillaire blanchâtre, entouré d’hémorragies péripapillaires. Elle survient comme le glaucome chez des patients âgés ; l’existence de baisse visuelle ou de diplopie, toutes deux transitoires, d’une altération de l’état général, d’un syndrome inflammatoire biologique et d’un retard choroïdien massif et prolongé permettent d’orienter le diagnostic qui sera confirmé par la biopsie d’artère temporale. Au stade tardif d’une NOIA artéritique, la papille prend un aspect pâle et une excavation est noté dans plus de 80 % des cas alors qu’elle est exceptionnelle dans la forme non artéritique [5, 6, 13, 16, 24, 27]. Les principaux facteurs qui contribuent à excaver le disque optique dans la NOIA artéritique sont l’importance de la perte en fibres visuelles liée à la thrombose des artères ciliaires courtes postérieures et la dégradation de la structure conjonctivale qui sert de support au disque avec une bascule postérieure de la lame criblée [12]. Les caractéristiques qui distinguent l’excavation de la NOIA artéritique de celle du glaucome sont principalement la pâleur, localisée à l’excavation dans le glaucome et étendue à tout le disque dans la NOIA artéritique ; pour une excavation de même importance, on ne retrouve pas de déficit focal de l’ANR dans la NOIA artéritique comme celle que l’on observe dans les disques glaucomateux (voir eCas clinique 14-3).
C’est une complication grave survenant après une procédure chirurgicale avec anesthésie générale. Elle a été décrite après tous les types de chirurgie mais principalement dans les interventions cardiaques et les chirurgies du rachis. Son incidence est plus grande chez les patients présentant des facteurs de risque vasculaire (diabète, HTA, artériosclérose), une maladie cardiaque, une insuffisance rénale, un glaucome à angle étroit, une collagénose ou une hémopathie. Les facteurs favorisants sont de longs temps opératoires, une hypotension artérielle prolongée, une anémie préexistante, une hémorragie digestive ou autre hémorragie peropératoire, la position couchée, voire une pression directe sur l’orbite et le globe. La position couchée et le Trendelenburg entraînent une diminution du retour veineux céphalique et une augmentation de la pression intracrânienne, ce qui contribue à majorer la pression au niveau du nerf optique ; la position couchée sur le ventre peut entraîner des blessures par compression directe au niveau de l’orbite. Au stade aigu le patient se réveille avec une baisse visuelle uni- ou bilatérale ; il s’agit le plus souvent d’une neuropathie optique ischémique antérieure, mais des neuropathies postérieures ont été décrites. Au stade d’atrophie optique, une excavation du disque optique semblable à celle de la NOIA artéritique avec pâleur de l’ANR s’installe [4].
C’est LE diagnostic à ne pas méconnaître car ces pathologies peuvent souvent bénéficier d’un traitement chirurgical et la précocité du diagnostic conditionne le résultat final sur la fonction visuelle. Ainsi, chez un patient qui consulte pour une baisse visuelle, la découverte d’une papille pâle et excavée, alors que la PIO rapportée à la mesure de la pachymétrie est normale et que l’histoire clinique et le champ visuel n’orientent pas d’emblée vers un mécanisme précis (vasculaire, héréditaire, toxique), doit faire pratiquer une imagerie (IRM) cérébrale et orbitaire injectée avec des coupes fines sur les voies visuelles antérieures afin d’éliminer une compression. Une compression doit également être éliminée s’il existe des signes associés à l’anomalie papillaire qui orientent vers une compression : exophtalmie, trouble oculomoteur, hypoesthésie dans le territoire du V1. De nombreuses publications ont décrit des lésions comprimant le nerf optique et s’accompagnant d’une excavation papillaire : compression orbitaire ou intracrânienne par un méningiome, une lésion suprasellaire ou un anévrisme intracrânien [17, 28]. Les patients présentant une neuropathie optique compressive sont plus jeunes que les patients glaucomateux et la baisse visuelle est souvent au premier plan. L’aspect du disque optique est variable et la pâleur, souvent marquée, peut être diffuse ou sectorielle (eCas cliniques 14-6 et 14-7).
Madame J., 25 ans, consulte pour une baisse visuelle gauche progressive, isolée et indolore. L’acuité est normale à droite, inférieure à 1/10 à gauche, et il existe un déficit pupillaire afférent relatif gauche. La papille droite est normale, la papille gauche est pâle, avec une pâleur prédominant en temporal (fig. A, B). Le champ visuel Octopus retrouve un large scotome central gauche (fig. C, D). L’IRM cérébrale et orbitaire en séquence T1 coupe coronale non injectée (fig. E) montre un gros nerf optique gauche. Le cliché en coupe sagittale avec suppression du signal de la graisse et injection de gadolinium retrouve un hypersignal péri- optique postérieur correspondant à un méningiome des gaines (fig. F).

A

B

C

D

E

F
Monsieur P., 47 ans, consulte pour une gêne visuelle bilatérale isolée. L’acuité est mesurée à 10/10 à droite et 8/10 à gauche. L’examen à la lampe à fente est normal, la PIO est mesurée à 14 ODG. Le fond d’œil retrouve une pâleur papillaire diffuse, avec une excavation à grand axe vertical en pente douce et un anneau atrophique péripapillaire (fig. A, B). Le champ visuel de Goldmann retrouve une tache aveugle exclue à droite (fig. C) et un déficit inférieur et temporal sans respect du méridien vertical à gauche (fig. D). L’IRM cérébrale montre un macro-adénome hypophysaire [séquence coronale T2 (fig. E) et sagittale T1 injectée (fig. F)] soulevant le chiasma optique.

A

B

C

D

E

F
La plupart des cas de NO traumatique sont associés à des accidents de la voie publique et une NO traumatique a été rapportée chez 3 % des traumatismes crâniens. En cas d’atteinte unilatérale, les patients présentent une baisse de l’acuité visuelle et un déficit pupillaire afférent relatif homolatéral. La cause la plus fréquente de NO traumatique est une atteinte du nerf dans la portion intracanalaire du nerf optique. En cas de traumatisme, les portions faiblement adhérentes du nerf optique se déplacent librement, ce qui induit une traction sur la partie intracanalaire avec une atteinte de la vascularisation et un cisaillement des axones. L’ensemble entraîne une perte axonale puis une excavation du disque et une pâleur.
Les NO toxiques au méthanol sont rares mais très sévères. L’intoxication aiguë peut avoir des conséquences graves avec une acidose métabolique et des troubles de la vigilance pouvant aller jusqu’au coma et au décès. Au stade aigu il existe un œdème papillaire blanchâtre qui est rapidement suivi d’une atrophie optique avec pâleur diffuse et excavation [19].
Les neuropathies optiques inflammatoires ou névrites optiques s’excavent rarement au stade atrophique. Elles posent peu de problèmes diagnostiques car le terrain (femme jeune), le mode d’installation rapidement progressif de la baisse visuelle unilatérale et douloureuse dans plus de 90 % des cas conduisent à demander une IRM cérébrale et orbitaire avec injection. Celle-ci affirme le diagnostic en mettant en évidence un hypersignal du nerf optique prenant le gadolinium et signant la rupture de la barrière hémato-encéphalique. L’existence d’autres hypersignaux sur l’IRM est un argument important pour une sclérose en plaques, étiologie principale des névrites optiques chez les patients caucasiens.
Les caractéristiques des neuropathies qui s’accompagnent d’une excavation papillaire et peuvent poser un problème diagnostique avec le GPN, ainsi que les points importants les concernant, sont résumés dans le tableau 14-7 [18].
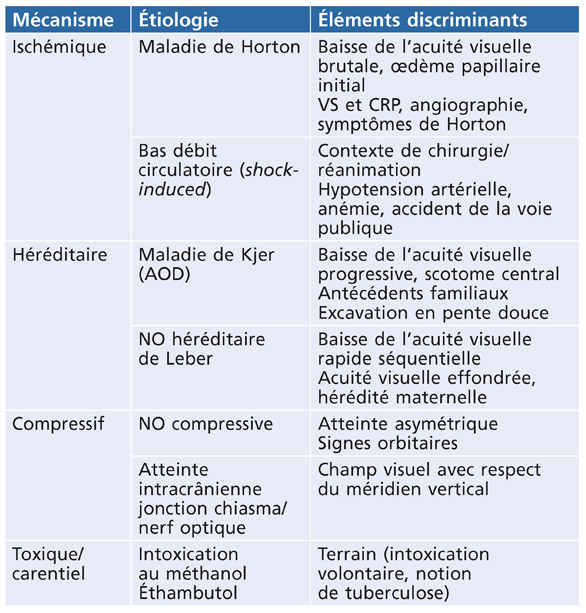
Tableau 14.7 – Caractéristiques des neuropathies s’accompagnant d’une excavation papillaire et pouvant poser un problème diagnostique avec le GPN.
Elles peuvent donner une pâleur et une excavation papillaire mais le champ visuel retrouve un déficit bitemporal respectant le méridien vertical, ce qui localise la lésion. Enfin, les atteintes des voies visuelles rétrochiasmatiques s’accompagnent d’un aspect normal des papilles et d’anomalies campimétriques, bilatérales, respectant le méridien vertical, ce qui n’est pas le cas du glaucome.
Quel bilan pratiquer devant une papille pâle et excavée ? Quand demander une imagerie cérébrale de complément ?
Plusieurs types de neuropathies optiques peuvent s’accompagner d’une modification de l’aspect de la papille simulant un glaucome. Le diagnostic se pose surtout lorsque la PIO est normale et repose sur l’analyse du profil clinique du patient et de la baisse visuelle, l’analyse du disque optique (clinique et OCT) et l’examen du champ visuel. Les indications de réalisation d’une IRM cérébrale et orbitaire figurent dans le tableau 14-8 [8].

Tableau 14.8 – Indications de réalisation d’une IRM cérébrale et orbitaire devant une papille pâle et excavée (d’après [20]).
Retenir
L’IRM cérébrale et orbitaire avec des coupes fines sur les voies visuelles permet de préciser le calibre et le signal des nerfs optiques et du chiasma et d’éliminer une compression. La NO glaucomateuse entraîne une atrophie progressive des nerfs optiques allant jusqu’au chiasma. L’IRM ne doit pas être systématique et sa demande doit être faite si l’on suspecte une atteinte non glaucomateuse [1].
Données de l’interrogatoire : l’existence d’une atteinte précoce de la vision centrale avec baisse d’acuité visuelle est un argument en faveur d’une NO non glaucomateuse, surtout si l’installation de la baisse visuelle a été brutale ou rapidement progressive, est unilatérale ou très asymétrique. Les circonstances d’installation de la baisse visuelle doivent être détaillées (traumatisme, chirurgie, toxique), de même que le terrain (âge, hérédité, facteurs de risque vasculaire, migraine, apnée du sommeil).
Données de l’examen clinique : la mesure de la PIO doit toujours être couplée à une pachymétrie et l’examen de l’angle iridocornéen être systématique. Une papille où la pâleur domine, et diffuse, alors que l’excavation même marquée est plutôt ronde et l’ANR régulier, est la conséquence d’une NO non glaucomateuse ou d’une pathologie chiasmatique.
Aspect du champ visuel : un déficit bitemporal respectant le méridien vertical doit faire éliminer une anomalie chiasmatique.
L’OCT papillaire avec étude du RNFL oriente vers un glaucome lorsque la perte en fibres optiques se localise en supérieur et/ou inférieur de la papille alors que le quadrant temporal est normal. Un déficit diffus d’emblée ou temporal oriente vers une atteinte non glaucomateuse.
[1] Ahmed II, Feldman F, Kucharczyk W, Trope GE. Neuroradiologic screening in normal-pressure glaucoma : study results and literature review. J Glaucoma. 2002 ; 11 : 279-86.
[2] Alward WL. The OPA1 gene and optic neuropathy. Br J Ophthalmol. 2003 ; 87 : 2-3.
[3] Ambati BK, Rizzo JF. Nonglaucomatous cupping of the optic disc. Int Ophthalmol Clin. 2001 ; 41 : 139-49.
[4] Berg KT, Harrison AH, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010 ; 4 : 531-46.
[5] Danesh-Meyer H, Savino PJ, Sergott RC. The prevalence of cupping in end-stage arteritic and nonarteritic anterior ischemic optic neuropathy. Ophthalmology. 2001 ; 108 : 593-8.
[6] Danesh-Meyer H, Savino PJ, Spaeth GL, Gamble GD. Comparison of arteritis and nonarteritic anterior ischemic optic neuropathies with the Heidelberg Retina Tomograph. Ophthalmology. 2005 ; 112 : 1104-12.
[7] Dutton GN. Congenital disorders of the optic nerve : excavations and hypoplasia. Eye. 2004 ; 18 : 1038-48.
[8] Girkin CA. The role of neuroimaging studies in normal-pressure glaucoma. J Glaucoma. 2002 ; 11 : 277-8.
[9] Greenfield DS, Siatkowski RM, Glaser JS, et al. The cupped disc : who needs neuroimaging ? Ophthalmology. 1998 ; 105 : 1866-74.
[10] Greenfield DS. Glaucomatous versus nonglaucomatous optic disc cupping : clinical differentiation. Semin Ophthalmol. 1999 ; 14 : 95-108.
[11] Gupta PK, Asrani S, Freedman SF, El-Dairi M, et al. Differentiating glaucomatous from non-glaucomatous optic nerve cupping by optical coherence tomography. The Open Neurology Journal. 2011 ; 5 : 1-7.
[12] Hayreh SS. Pathogenesis of cupping of the optic disc. Br J Ophthalmol. 1974 ; 58 : 863-76.
[13] Hayreh SS, Jonas JB. Optic disc morphology after arteritic anterior ischemic optic neuropathy. Ophthalmology. 2001 ; 108 : 1586-94.
[14] Jacobson L, Hellström A, Flodmark O. Large cups in normal-sized optic discs : a variant of optic nerve hypoplasia in children with periventricular leukomalacia. Arch Ophthalmol. 1997 ; 114 : 1263-9.
[15] Jonas JB, Budde WM, Panda-Jonas S. Ophthalmoscopic evaluation of the optic nerve head. Surv Ophthalmol. 1999 ; 43 : 293-320.
[16] Jonas JB, Hayreh SH, Tao Y, et al. Optic nerve head change in non-arteritic anterior ischemic optic neuropathy and its influence on visual outcome. PloS One. 2012 ; 7 : e37499.
[17] Kupersmith MJ, Krohn D. Cupping of the optic disc with compressive lesions of the anterior visual pathway. Ann Ophthalmol. 1984 ; 16 : 948-53.
[18] Lamirel C. Glaucome à pression normale. Quand suspecter une neuropathie non glaucomateuse ? Quand demander des examens complémentaires ? Les Cahiers d’Ophtalmologie. 2013 ; 169 : 24-28.
[19] Lloyd MJ, Fraunfelder FW. Drug-induced optic neuropathies. Drugs Today. 2007 ; 43 : 827-36.
[20] Milea D. Excavation papillaire non glaucomateuse. J Fr Ophthalmol. 2007 ; 30 : 3s31-3s34
[21] Moore M, Salles D, Jampol LM. Progressive optic nerve cupping and neural rim decrease in a patient with bilateral autosomal dominant optic nerve colobomas. Am J Ophthalmol. 2000 ; 129 : 517-20.
[22] Morgan JE, Sheen NJ, North RV, et al. Discrimination of glaucomatous optic neuropathy by digital stereoscopic analysis. Ophthalmology. 2005 ; 112 : 855-62.
[23] O’Neill EC, Danesh-Meyer HV, Kong GX, et al. Optic Nerve Study Group. Optic disc evaluation in optic neuropathies : the optic disc assessment project. Ophthalmology. 2011 ; 118 : 964-70.
[24] Orgül S, Gass A, Flammer J. Optic disc cupping in arteritic AION. Ophthalmologica. 1994 ; 208 : 336-8.
[25] Ortiz RG, Newman NJ, Manoukian SV, et al. Optic disk cupping and electro-cardiographic abnormalities in an American pedigree with Leber’s hereditary optic neuropathy. Am J Ophthalmol. 1992 ; 113 : 561-6.
[26] Piette SD, Sergott RC. Pathological optic-disc cupping. Curr Opin Ophthalmol. 2006 ; 17 : 1-6.
[27] Quigley H, Anderson DR. Cupping of the optic disc in ischemic optic neuropathy. Trans Am Acad Ophthalmol Otol. 1977 ; 83 : 755-62.
[28] Stewart WC, Reid KK. Incidence of systemic and ocular disease that may mimic low-tension glaucoma. J Glaucoma. 1992 ; 1 : 27-31.
[29] Trobe JD, Glaser JS, Cassady JC, et al. Non glaucomatous excavation of the optic disc. Arch Ophthalmol. 1980 ; 98 : 1046-50.
[30] Trobe JD, Glaser JS, Cassady JC. Optic atrophy. Differential diagnosis by fundus observation alone. Arch Ophthalmol. 1980 ; 98 : 1040-5.
[31] Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol. 2003 ; 87 : 48-53.
- Chapitre 14
Diagnostic différentiel