

A. Denoyer
« Le simple est toujours faux. Ce qui ne l’est pas est inutile. »
Paul Valéry, Mauvaises pensées et autres (1942)
La mort par apoptose des cellules ganglionnaires rétiniennes joue un rôle central au sein des mécanismes impliqués dans la neurodégénérescence rétinienne glaucomateuse.
Certaines caractéristiques intrinsèques de la cellule ganglionnaire rétinienne conditionnent sa sensibilité aux voies membranaires et mitochondriales de l’apoptose.
En miroir, quatre facteurs majeurs d’agression favorisent la perte neuronale : la carence en neurotrophines, l’excitotoxicité du glutamate, l’ischémie et le stress oxydatif, et l’activation gliale.
Il existe en parallèle un processus d’autodestruction axonale et dendritique non apoptotique conduisant à une désorganisation structurale et fonctionnelle de la neurorétine glaucomateuse.
Seule une approche globale des interactions cellulaires et des mécanismes impliqués dans la neurodégénérescence glaucomateuse permettra de développer une véritable stratégie thérapeutique neuroprotectrice qui fait encore défaut aujourd’hui.
Le glaucome constitue un groupe spécifique au sein des neuropathies optiques chroniques caractérisé par une neurodégénérescence rétinienne associée à la disparition progressive des cellules ganglionnaires rétiniennes (CGR). La neuropathie glaucomateuse est une pathologie complexe, génétique et multifactorielle dont les mécanismes sous-jacents demeurent aujourd’hui très mal compris. Ainsi, l’élévation de la pression intra-oculaire (PIO), premier facteur de risque connu – mais non indispensable –, demeure à ce jour le seul événement pathogénique accessible aux traitements médicaux ou chirurgicaux [2]. La neurorétine semble pourtant être le terrain de modifications pathologiques en réaction directe à l’agression hypertensive extrinsèque d’une part, mais aussi de phénomènes secondaires bénéfiques ou délétères liés à ses caractéristiques intrinsèques d’autre part. Naturellement, l’amélioration de nos connaissances sur les mécanismes impliqués dans la neuropathie glaucomateuse ainsi qu’une appréhension de la pathologie dans sa globalité pourraient ouvrir la voie de nouvelles stratégies thérapeutiques ciblant spécifiquement les processus neurodégénératifs mis en jeu. L’idée de neuroprotection dans le glaucome n’est pas récente, mais son application en pratique clinique ne s’est pour l’instant soldée que par des échecs [23]. La communauté scientifique en charge de la recherche dans le glaucome se heurte à certains écueils, fondamentaux pour part comme la diversité des acteurs cellulaires et de leurs interactions ou bien la pluralité des polymorphismes géniques, mais aussi plus pratiques comme l’absence de modèles cellulaires ou animaux reproduisant de façon sensée la pathologie.
L’approche détaillée des mécanismes physiopathologiques impliqués dans la neuropathie glaucomateuse peut se révéler complexe et fastidieuse pour le clinicien. Celle-ci permet cependant de replacer la pathologie dans son ensemble et participe certainement à la mise en perspective des échecs cliniques, que nous rencontrons parfois, vers un avenir thérapeutique plus satisfaisant [7]. Ce chapitre unifie donc les différentes pistes physiopathologiques connues et moins connues dans une approche schématique, hiérarchisée et critique qui tente d’éviter les écueils d’une simple liste exhaustive qui serait dépourvue de sens.
Au cours de la neuropathie glaucomateuse, les CGR meurent essentiellement par apoptose [19], un processus létal constitué par une cascade d’événements intracellulaires assimilable à une séquence d’autodestruction de la cellule. L’apoptose est caractérisée par des altérations morphofonctionnelles spécifiques comme la condensation de la chromatine, la fragmentation précise de l’ADN et l’altération structurelle des membranes cytoplasmiques et mitochondriales. Les caractéristiques qui la différencient de la nécrose sont résumées dans le tableau 8-1.
Schématiquement, deux voies essentielles, la voie extrinsèque et la voie mitochondriale, en relation avec le rôle clé des flux cellulaires du calcium et de l’adénosine triphosphate (ATP), conduisent à un tel phénomène neurodégénératif, mais ces différentes voies sont cependant intimement liées. La figure 8-1 intègre la complexité des processus mis en jeu.

Tableau 8.1 – Quels éléments caractérisent la mort cellulaire par apoptose ? Résumé des différences qui opposent apoptose et nécrose.

Fig. 8-1 Principales voies d’apoptose sollicitées au cours de la mort des cellules ganglionnaires rétiniennes. Aux voies extrinsèque (voie des récepteurs membranaires) et intrinsèque mitochondriale s’associent les effets directs des flux de calcium et d’ATP.
La voie dite extrinsèque regroupe les récepteurs pro-apoptotiques de la membrane cytoplasmique au premier rang desquels figurent les récepteurs du TNF-α : TNF-R1 et TNF-R2 (TNF : tumor necrosis factor). Le TNF-α est une cytokine qui joue un rôle fondamental dans l’immunité, l’inflammation et le contrôle de la viabilité cellulaire. Sa liaison au récepteur TNF-R1 a un effet pro-inflammatoire et pro-apoptotique, alors que l’activation du TNF-R2 influencerait préférentiellement la prolifération cellulaire, le rôle de ce dernier demeurant encore aujourd’hui sujet à controverses. L’activation du récepteur TNF-R1 entraîne essentiellement une apoptose par la voie des caspases, en activant schématiquement la caspase 8 puis la caspase 3 qui « digèrent » alors directement les constituants du noyau cellulaire et induisent une apoptose. Un second mode d’action, dit caspase-indépendant, serait lié à l’induction de canaux cytoplasmiques perméables au calcium (Ca-perméable AMPAR) responsables d’un afflux intracellulaire de calcium – ses conséquences délétères sont détaillées plus loin. Enfin, l’activation des TNF-R déclenche un certain nombre de mitogen-activated protein kinases (MAPK) dont les effets protecteurs ou délétères sont détaillés plus loin.
Dans le glaucome, le TNF-α semble occuper aujourd’hui un rôle central. Il a été montré une surexpression de TNF-α dans la rétine et le vitré non seulement dans des modèles expérimentaux de glaucome mais aussi chez les patients atteints de glaucome [34]. De plus, certains polymorphismes du gène du TNF-α seraient corrélés à une incidence accrue de glaucome [24]. Chez l’animal, la suppression de l’expression membranaire des récepteurs TNF-R ou bien la diminution de synthèse de TNF-α joue un rôle protecteur vis-à-vis des CGR. Au cours de la neuropathie glaucomateuse, il semblerait qu’une surexpression de TNF-α par les cellules gliales de Müller (CGM) contribue ainsi à la neurodégénérescence [5]. Notons enfin que l’inhibition combinée de l’activation de certaines caspases permettrait d’améliorer la survie des CGR dans des modèles animaux de glaucome.
Fas-ligand est aussi une cytokine qui présente une isoforme pro-apoptotique via son récepteur Fas/CD95. Le complexe Fas/Fas-ligand est impliqué dans certaines réactions inflammatoires ainsi que dans le contrôle de la prolifération ou l’apoptose cellulaire. Le récepteur Fas est exprimé à la surface des neurones et des cellules gliales comme les astrocytes et la microglie.
Dans la neuropathie glaucomateuse, il a été montré chez l’animal qu’une surexpression de Fas-ligand par la microglie était à l’origine d’une dégénérescence neuronale. De façon plus précise, il semble coexister deux isoformes, sFasL et mFasL, aux propriétés protectrices pour la première et délétères pour la seconde, leur équilibre pouvant être impliqué dans la régulation de la viabilité neuronale [14].
Les MAPK constituent une des voies essentielles de signalisation intracellulaire et relaient une grande variété de fonctions cellulaires. Les deux principales familles de MAPK impliquées dans l’apoptose neuronale sont les Janus kinases (JNK) qui activent le facteur de transcription C-jun et les différentes isoformes de la p38. Expérimentalement, une surexpression de la voie JNK-jun ou bien de certaines isoformes de p38 a été rapportée dans des modèles de neuropathie traumatique et des modèles d’hypertonie intra-oculaire, et la suppression de ces voies de signalisation assure une neuroprotection des CGR. Chez l’homme, une étude a rapporté une suractivité de la voie JNK-jun en cas de glaucome [37].
En tout état de cause, les MAPK sont impliquées dans la régulation de la viabilité des CGR. La multiplicité de leurs isoformes et la pluralité de leurs fonctions sont néanmoins à l’origine de difficultés majeures dans l’étude de leur rôle spécifique au cours du glaucome, et le ciblage ultra-spécifique d’une de ces voies à visée thérapeutique semble peu envisageable aujourd’hui.
La mitochondrie assure la majorité de la chaîne respiratoire cellulaire, fournissant aux neurones la quantité d’énergie nécessaire à leur activité. La grande famille Bcl-2 constitue un ensemble de protéines qui contrôlent la perméabilité de la membrane mitochondriale. Schématiquement, Bcl-2 et Bcl-XL sont impliquées dans la survie neuronale alors que Bax et les protéines BH3 (BAD, BID, BIM) jouent un rôle central dans l’apoptose. Toutes ces protéines sont exprimées dans les CGR et pourraient donc moduler ou relayer les phénomènes neurodégénératifs au cours du glaucome [27], mais un lien direct entre glaucome et Bcl-2 n’a jamais été clairement démontré.
La perméabilisation pathologique de la membrane mitochondriale libère certaines protéines pro-apoptotiques, au premier rang desquelles figure le cytochrome c. En excès dans le cytoplasme, celui-ci forme un complexe nommé apoptosome qui à son tour activera la voie des caspases via la caspase 9. D’autres protéines d’origine mitochondriale participent à l’apoptose cellulaire, en particulier les protéines DIABLO, l’AIF et HTRA2/OMI.
Les contraintes mécaniques (élévation de la PIO), hémodynamiques (ischémie de la tête du nerf optique) et oxydatives rencontrées de façon pathologique au sein du tissu neurorétinien glaucomateux pourraient ainsi déréguler la fonction mitochondriale, fragiliser les CGR en termes de fonction neuronale et induire une apoptose [28]. Ainsi, certaines altérations géniques mitochondriales mises en évidence chez des patients atteints de glaucome primitif à angle aigu (GPAO) pourraient contribuer à la dégénérescence neurorétinienne. Enfin, la diminution de la synthèse d’ATP secondaire aux dysfonctions mitochondriales pourrait aussi contribuer à cette dégénérescence [46] (voir plus loin).
Le calcium joue un rôle clé dans la signalisation cellulaire et neuronale dans l’organisme. Il a été montré chez l’animal qu’un stress du nerf optique entraînait un influx majeur de calcium en provenance du milieu extracellulaire et du réticulum endoplasmique, associé à une neurodégénérescence [43]. L’afflux rapide de calcium intracellulaire favorise la synthèse de monoxyde d’azote et d’espèces réactives de l’oxygène responsables d’un stress oxydatif contribuant à l’apoptose (voir plus loin). En outre, le calcium active certaines protéines comme la caspaïne et la calcineurine, facteurs pro-apoptotiques directement, par l’intermédiaire des caspases et des protéines de perméabilisation mitochondriales, essentiellement BAD [15].
Dans le glaucome, il a été montré qu’un influx pathologique de calcium était associé à la dégénérescence neuronale rétinienne [30]. Les causes de ce flux sont multiples et débattues : citons la défaillance de certaines pompes ioniques (Na+/K+-ATPase), l’ouverture de pores perméables au calcium (AMPAR) ou encore l’excitotoxicité de la voie du glutamate via les récepteurs NMDA (voir plus loin) [42].
L’ATP assure la transmission de l’énergie du milieu extracytoplasmique (mitochondrial et extracellulaire) vers le cytoplasme cellulaire, énergie indispensable à la viabilité et la fonction neuronale. Chez l’animal, l’hypertonie intra-oculaire, associée aussi à une dysfonction mitochondriale, entraînerait d’une part une diminution de l’ATP intracytoplasmique et d’autre part une dérégulation de son transport axonal, déclenchant ainsi une apoptose des CGR [6, 8].
L’autre versant de l’action pro-apoptotique de l’ATP est lié à sa toxicité en cas d’excès dans le milieu externe extracellulaire. Pour une CGR donnée, la mort d’autres cellules rétiniennes dans son environnement proche entraîne un excès d’ATP extracellulaire qui active les récepteurs purinergiques, essentiellement P2X, et déclenche une apoptose. Il a été ainsi montré un excès d’ATP dans l’humeur aqueuse de patients atteints de glaucome aigu par fermeture de l’angle (GAFA) [46]. Chez l’animal, le contrôle des mouvements d’ATP par l’administration de coenzyme Q10 ou le blocage des pores P2X auraient un effet protecteur en cas de stress des CGR [25].
Comme dans tout processus de mort cellulaire, la dégénérescence des CGR résulte d’un déséquilibre entre les facteurs intrinsèques et extrinsèques de protection et les signaux pro-apoptotiques. La figure 8-2 modélise les principaux acteurs qui contribuent à l’apoptose neuronale glaucomateuse.
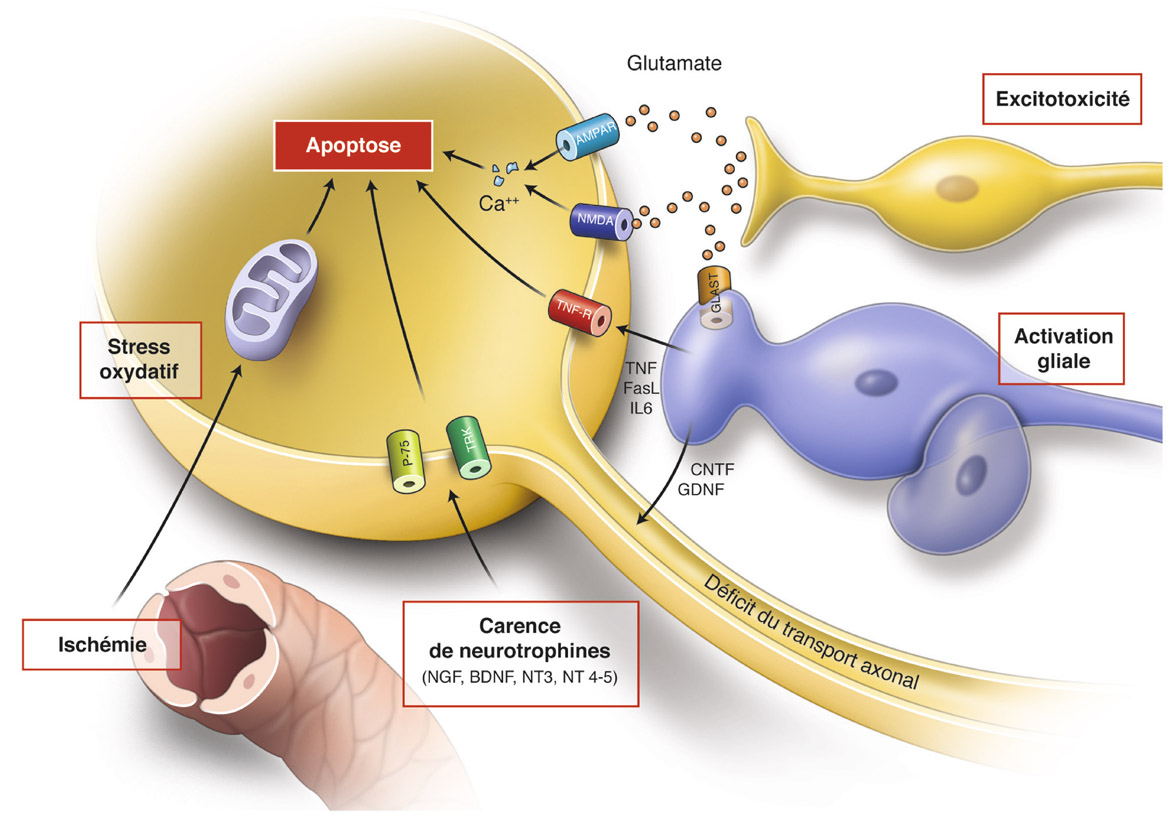
Fig. 8-2 Facteurs d’agression de la CGR au cours de la neuropathie glaucomateuse. Ischémie/stress oxydatif, carence en neurotrophines, excitotoxicité du glutamate et activation gliale sont les quatre principaux éléments favorisant la neurodégénérescence de la CGR.
Les facteurs neurotrophiques, ou neurotrophines, sont des protéines de bas poids moléculaire qui régulent le développement, la fonction et la viabilité des neurones.
Les quatre principales neurotrophines sont :
le nerve growth factor (NGF) ;
le brain-derived neurotrophic factor (BDNF) ;
la neurotrophine-3 (NT-3) ;
la neurotrophine-4/5 (NT-4/5).
Au niveau neuronal, les neurotrophines peuvent activer soit les récepteurs de la famille Trk, dont l’action semble plutôt protectrice via la protéine kinase Erk1/2, soit le récepteur p75NT, très similaire aux récepteurs TNF-R évoqués plus haut, et déclencher une cascade de signalisation favorisant généralement l’apoptose, essentiellement via JAK/STAT3.
Les neurotrophines peuvent agir de façon autocrine, c’est-à-dire directement sécrétées et recaptées par la terminaison synaptique. Cette voie d’action minoritaire entraîne une carence en neurotrophines en cas de rétraction des terminaisons synaptiques, donc à un stade avancé de dégénérescence neuronale. En réalité, la majorité des neurotrophines est d’origine centrale (synthétisée au niveau du colliculus concernant les CGR) puis transportée de façon rétrograde le long de l’axone jusqu’à la terminaison nerveuse. Dans ce cas, c’est donc un déficit du transport axonal rétrograde qui est à l’origine d’une privation en facteurs neurotrophiques [3].
Dans le cadre de la neuropathie glaucomateuse, il a été montré chez l’animal – mais aussi chez l’homme dans le cadre de glaucomes secondaires – que l’hypertonie oculaire était à l’origine d’une défaillance du transport axonal antérograde et rétrograde entraînant une carence en BDNF [32]. Ainsi, l’apport complémentaire de BDNF par injection intravitréenne ou transfection de vecteurs viraux améliorerait la morphologie, la fonction et la survie des CGR dans des modèles animaux de neuropathie [12]. Plus récemment, il a été montré que l’apport exogène de NGF par voie topique réduirait l’apoptose neuronale et améliorerait la fonction visuelle des patients glaucomateux [20]. Cependant, la complexité des voies de signalisation conditionnant l’équilibre précaire entre viabilité et apoptose des CGR en réponse aux neurotrophines d’une part, et la variabilité d’expression des récepteurs Trk à la surface des CGR d’autre part, semble faire obstacle à la généralisation de tels procédés thérapeutiques aujourd’hui. Les principales voies de recherche se concentrent essentiellement sur le couple BDNF/Trk, les autres axes neurotrophiques ne semblant à ce jour pas assez connus pour bénéficier d’une approche thérapeutique spécifique.
Dans la rétine, deux autres facteurs neurotrophiques ont été identifiés comme acteurs potentiels de l’apoptose des CGR au cours du glaucome : le ciliary neurotrophic factor (CNTF) et le glial cell line-derived neurotrophic factor (GDNF).
Le CNTF semble essentiellement synthétisé par les cellules gliales de Müller, particulièrement dans des modèles animaux d’axonotomie, d’ischémie et d’hypertonie intra-oculaire [44]. Le CNTF des cellules de Müller se lierait ainsi aux récepteurs CNTF-α très exprimés à la surface des CGR, favorisant la survie neuronale via les kinases Erk1/2, JAK/STAT3 et PI3K. Les tentatives d’apport de CNTF exogène, essentiellement par transfection virale, ont permis de protéger la neurorétine dans des modèles animaux d’ischémie et d’hypertonie, voire de favoriser la repousse axonale, ce qui différencierait le CNTF des autres neurotrophines [21]. On pourrait ainsi imaginer à l’avenir une thérapie combinant BDNF et CNTF favorisant de façon complémentaire la survie des CGR et la repousse axonale respectivement. Enfin, le GDNF aurait des propriétés comparables à celles du CTNF. Les astrocytes et les neurones seraient à l’origine d’une synthèse de GDNF qui activerait les récepteurs GFRα1 et tkRET au niveau de la synapse.
L’excitotoxicité désigne l’effet délétère direct du principal facteur de neurotransmission au sein de la rétine : le glutamate. Le glutamate participe à la neurotransmission d’une part entre les photorécepteurs et les cellules bipolaires, et d’autre part entre les cellules bipolaires et les CGR. Son recaptage neuronal s’effectue essentiellement via les récepteurs NMDA. En cas d’excès extracellulaire de glutamate, l’activation des récepteurs NMDA entraîne un afflux massif de calcium intracellulaire dont les effets délétères ont été préalablement détaillés. L’effet apoptotique puissant du glutamate sur les CGR a été largement démontré chez l’homme in vitro et in vivo [3, 22]. Certaines études ont rapporté une élévation pathologique de la concentration extracellulaire de glutamate chez l’animal ainsi que chez des patients atteints de GPAO [13] ; d’autres ne l’ont pas identifiée. Enfin, les cellules gliales de Müller expriment aussi les récepteurs NMDA dont l’activation induit la synthèse de TNF-α toxique pour les CGR. Pour résumer, il semblerait que des variations récurrentes de concentration synaptique du glutamate puissent être impliquées dans la neurodégénérescence glaucomateuse, mais l’utilisation thérapeutique d’antagonistes des récepteurs NMDA s’est toujours soldée par un échec [29].
Il existe une grande variété de transporteurs non NMDA du glutamate, en particulier GLAST (glutamate aspartate transporter), chargés de capter le glutamate extracellulaire en excès au niveau synaptique. Ces transporteurs sont largement exprimés par les neurones, mais aussi par les cellules gliales qui semblent jouer un rôle essentiel dans la régulation de l’excitotoxicité du glutamate. Chez l’animal, le déficit en GLAST exacerberait la dégénérescence neuronale en cas d’hypertension oculaire, mais les mécanismes de régulation sous-jacents demeurent encore inconnus.
Enfin, les calcium-dépendants AMPAR (voir plus haut) et AMPA-KR constitueraient aussi une voie de signalisation « non NMDA » du glutamate responsable d’apoptose neuronale. Leur surexpression a été rapportée dans de nombreux modèles d’ischémie, d’excitotoxicité et de processus neurodégénératifs. Leurs rôles spécifiques dans le glaucome sont inconnus à ce jour.
Le stress oxydatif est la conséquence d’un excès d’espèces réactives de l’oxygène (ROS, reactive oxygen species) secondaire à une dysfonction mitochondriale ou bien à la dégradation enzymatique de nombreuses protéines. Les ROS dégradent les protéines et l’ADN, entraînant ainsi une mort cellulaire. Dans le glaucome, l’hypertonie oculaire associée aux troubles de perfusion du nerf optique pourrait contribuer au stress oxydatif rétinien. Certaines ROS ainsi que les dommages intracellulaires qu’elles engendrent ont été retrouvés chez l’animal et chez les patients atteints de GPAO [16]. En réaction à l’hypoxie, la mitochondrie est à l’origine d’une cascade de transcription génique via HIF-1 aboutissant en particulier à la synthèse d’érythropoïétine, de VGF et de la protéine Hsp-27, pour lesquels des concentrations élevées ont été retrouvées dans l’humeur aqueuse de patients glaucomateux [38].
La cellule dispose en outre de nombreux mécanismes anti-oxydants dont la dysfonction pourrait aussi participer à la neurodégénérescence rétinienne. Ainsi, certaines protéines anti-oxydantes comme la superoxyde dismutase, la catalase, la glutathione peroxydase et la glutathione réductase semblent impliquées dans l’apoptose des CGR chez l’animal. Néanmoins, aucune piste thérapeutique spécifique n’a encore émergé de cette voie.
Les astrocytes, qui font partie de la macroglie, sont en étroite relation avec les CGR avec lesquelles ils entretiennent des rapports biologiques et mécaniques privilégiés [47]. Dans le GPAO, trois modifications astrocytaires pathologiques ont été décrites chez l’animal et chez l’homme au niveau de lame criblée [17] :
une prolifération et une hypertrophie cellulaires (astrocytose) ;
l’expression de marqueurs d’activation comme la GFAP (glial fibrillary acidic protein) ;
une dérégulation de l’expression de certains gènes (plus précisément la surexpression de MMP [métalloprotéases matricielles] et de certains composants de la matrice extracellulaire).
L’ensemble de ces modifications astrocytaires contribuerait au remodelage pathologique de la tête du nerf optique responsable d’une compression axonale directe ainsi que d’une ischémie rétinienne liée à la compression vasculaire. Notons que ces altérations astrocytaires sont retrouvées essentiellement au niveau de la tête du nerf optique, et non pas dans l’ensemble du tissu rétinien, ce qui plaide en faveur de l’existence d’une population astrocytaire individualisée au niveau du nerf optique dont le comportement spécifique conditionnerait l’apoptose glaucomateuse [31].
Enfin, les astrocytes sont aussi sensibles à certains composés et protéines retrouvés en excès au cours du glaucome : le NO, certaines interleukines, le TNF-α et les endothélines. Par exemple, l’excès d’endothéline-1 chez l’animal entraînerait une ischémie et une mort neuronale liée à l’expression membranaire astrocytaire de récepteurs aux endothélines [11].
Les cellules gliales de Müller sont les principales actrices de la régulation de l’homéostasie rétinienne [9]. En conditions physiologiques, elles assurent une protection neuronale en régulant les flux d’eau et d’ions, en participant au recyclage du glutamate via la glutamine synthétase, à la production d’anti-oxydants et la production de neurotrophines (CNTF).
Dans le glaucome, les cellules gliales de Müller réagissent très rapidement à l’hypertension intra-oculaire qui induit une activation cellulaire (hypertrophie, expression de GFAP, activation de la voie ERK1/2), une dérégulation des fonctions protectrices physiologiques, voire la synthèse de facteurs neurotoxiques comme le NO, le TNF-α et l’α2-macroglobine [18]. L’ensemble de ces dysfonctions participerait à l’apoptose neuronale précoce ainsi qu’à la dysfonction neuronale. Des travaux inédits de notre équipe semblent montrer que l’OCT-SD (spectral domain) serait en mesure de dépister spécifiquement de telles altérations morphologiques et que celles-ci seraient corrélées à la dysfonction visuelle (voir encadré et fig. 8-3). Enfin, certaines équipes de recherche explorent le rôle non démontré des cellules de Müller dans le remplacement neuronal par transdifférenciation [10].
La microglie est l’ensemble des cellules immunitaires qui résident de façon permanente dans le tissu rétinien. Ces cellules macrophagiques assurent essentiellement un rôle de protection immunitaire, d’élimination des débris cellulaires et de synthèse de facteurs neurotrophiques nécessaires à la survie des CGR.
Une activation microgliale a été détectée chez l’homme et dans des modèles animaux de glaucome [45]. Tout comme l’activation astrocytaire, celle-ci entraînerait une synthèse pathologique de facteurs neurotoxiques – NO, interleukines, TNF-α et endothélines –, ainsi qu’un défaut de clairance des débris cellulaires et radicaux libres. L’inhibition ciblée de l’activation microgliale chez l’animal diminue la mort neuronale dans des modèles animaux de glaucome, constituant ainsi une piste thérapeutique prometteuse.
De nombreuses preuves s’accumulent en faveur d’une désorganisation des connexions synaptiques rétiniennes au cours de la neuropathie glaucomateuse. Chez l’animal, il semblerait qu’une hypotrophie associée à une perte des ramifications dendritiques des CGR survienne bien avant la perte axonale [41]. Cette désorganisation de l’arborescence dendritique serait associée d’une part à une diminution de la connectivité neuronale à l’échelle cellulaire (sous-expression de c-fos, un marqueur de communication intercellulaire), et d’autre part à une diminution de la réponse spatiale et temporelle à un stimulus visuel sur le plan fonctionnel [40].
Certaines neurotrophines pourraient ainsi participer au remodelage de l’arborescence dendritique des CGR. Le couple BDNF/TrkB et le CNTF déjà mentionnés plus haut pourraient ainsi participer à la régulation de la connectivité synaptique. Des études dans des modèles de neuropathie par lésion axonale plaideraient en faveur d’un rôle protecteur de ces neurotrophines sur l’organisation dendritique des CGR. En parallèle, certaines fractions du complément (C1q, C3, C4 et C5) pourraient au niveau synaptique participer à l’organisation des dendrites et des connexions neuronales [36]. Des études récentes ont montré que l’excès de C1q dans des modèles de glaucome et d’ischémie pourrait contribuer à une désorganisation de l’arborescence neuronale et participer à des troubles fonctionnels précoces suivis d’une apoptose secondaire [33]. Ainsi, la voie du complément pourrait aussi constituer une cible thérapeutique de neuroprotection.
En miroir de la désorganisation dendritique rétinienne, les connexions synaptiques des CGR au niveau du corps genouillé latéral seraient aussi altérées au cours de la neuropathie glaucomateuse [39]. Chez l’homme, un appauvrissement de l’arborescence dendritique ainsi qu’une atrophie et une apoptose neuronales ont été retrouvés dans les corps genouillés latéraux de patients atteints de glaucome, suggérant ainsi que la neuropathie glaucomateuse se prolonge bien au-delà de l’œil, dans le système nerveux central.

Fig. 8-3 Anomalies maculaires glaucomateuses détectées par tomographie en cohérence optique (OCT) et corrélations morphofonctionnelles
(Collection A. El Maftouhi ; d’après El Maftouhi A, et al. Inner nuclear layer cystic degeneration, in the macular area of glaucomatous patients, on En face and B scan OCT. In Clinical En Face OCT Atlas. Jaypee-Highlights Medical Publishers Inc., 2013.)
a. OCT en face et en coupes d’anomalies microkystiques maculaires situées au sein de la couche nucléaire interne. La forme – en fuseaux perpendiculaires au plan rétinien – et la localisation sembleraient en faveur d’une anomalie des cellules gliales de Müller, sans pouvoir déterminer si l’épaississement de la nucléaire interne est lié à une ballonisation cellulaire ou bien un excès de fluides extracellulaires.
b. Les microkystes sont associés à un amincissement de la couche des cellules ganglionnaires qui correspond exactement au déficit visuel central. Cette correspondance morphofonctionnelle mise en évidence de façon inédite pourrait étayer l’hypothèse d’une gliose müllérienne précoce associée au dysfonctionnement puis à la dégénérescence des cellules ganglionnaires de voisinage.
Dans la majorité des types cellulaires, la mort ou bien la survie résultent d’un équilibre entre des signaux protecteurs et des signaux délétères, équilibre qui détermine le devenir de la cellule dans sa totalité. Les neurones constituent une population cellulaire d’exception dont l’hyper-spécialisation est associée à la capacité de compartimentaliser la réponse aux signaux, autrement dit de la restreindre à une seule partie de la cellule et non pas sa totalité. Ainsi, la CGR répond au stress par des processus dégénératifs spécifiques à l’axone, spécifiques aux dendrites et spécifiques à la synapse, bien avant que ne se produise une mort du corps – le soma – cellulaire [42].
Schématiquement, l’autodestruction axonale peut avoir lieu selon deux processus différents :
la dégénérescence wallérienne, qui correspond à une destruction rapide, antérograde et totale de l’axone et la terminaison synaptique suite à un stress aigu ;
la dégénérescence rétrograde, qui correspond à une destruction différée, rétrograde (qui débute par la terminaison synaptique et remonte le long de l’axone) et progressive en réponse à un stress chronique.
La dégénérescence wallérienne se rencontre dans les accidents vasculaires cérébraux, les traumatismes, les infections et la maladie d’Alzheimer, alors que la dégénérescence rétrograde est volontiers secondaire aux neuropathies toxiques et métaboliques périphériques ainsi qu’aux encéphalopathies liées au VIH. De la même façon, il existe aussi une autodestruction dendritique et une autodestruction synaptique qui peuvent avoir lieu de façon indépendante, sans apoptose neuronale, et ont été décrites dans de nombreux processus neurodégénératifs [35].
Ces processus d’autodestruction sont contrôlés essentiellement par le protéasome et par les flux cellulaires de calcium qui jouent un rôle clé dans la dégénérescence axonale. En revanche, ils semblent indépendants des mécanismes conventionnels d’apoptose : ni la famille Bcl2 ni les caspases ne seraient impliquées. En outre, chez les souris Wlds spontanément résistantes à la dégénérescence axonale, les neurones meurent par apoptose indépendamment de tout phénomène d’autodestruction. L’autodestruction axonale, dendritique et/ou synaptique pourrait donc se limiter à une altération non apoptotique d’une partie de la cellule, puis éventuellement engendrer une apoptose secondaire et différée.
Au cours du glaucome, il est probable que les CGR subissent un stress axonal prononcé au niveau de la lame criblée qui résulterait des contraintes mécaniques induites directement par l’hypertension oculaire, de l’ischémie locale et des modifications de l’environnement glial, essentiellement astrocytaire comme expliqué plus haut. Ce stress pourrait être responsable d’une dégénérescence synchrone de type wallérienne, indépendante de l’apoptose, qui expliquerait la raréfaction axonale et entraînerait secondairement une atrophie somatique, puis une mort neuronale [4]. En outre, la carence en facteurs neurotrophiques par dégradation du transport axonal [32] et l’exposition des CGR à certaines cytokines pourraient exercer une contrainte micro-environnementale chronique responsable d’une autodestruction axonale asynchrone de type rétrograde.
L’hypothèse d’une autodestruction dendritique a aussi été évoquée au niveau rétinien (voir plus haut). De rares travaux ont révélé la présence d’anomalies dendritiques – amincissement, formation de bulles, appauvrissement de l’arborescence – et synaptiques dans des modèles animaux de glaucome. Ces éléments encore mal connus sont d’une importance cruciale car ils pourraient non seulement être associés à une perte fonctionnelle précoce mais aussi déterminer une apoptose neuronale secondaire, à retardement.
Cette notion émergente de compartimentalisation de la réponse neuronale laisse entrevoir la complexité des processus qui sous-tendent la dégénérescence rétinienne glaucomateuse et la nécessité d’envisager la mort des CGR non seulement cellule par cellule, mais aussi compartiment cellulaire par compartiment cellulaire. En outre, ces phénomènes de dégénérescence spécifiques aux neurones pourraient expliquer pour part les raisons des multiples échecs des thérapies neuroprotectrices [1, 7].
La diversité des intervenants évoqués tout au long de ce chapitre révèle la complexité des mécanismes impliqués dans la neuropathie glaucomateuse. Mais plus encore que le nombre et le rôle de chacun de ces acteurs, les relations de causalité et de temporalité qui les lient les uns aux autres restent non élucidées. Néanmoins, en regroupant l’ensemble des connaissances actuelles, il semble possible de décrire une séquence d’événements qui conduirait au glaucome tel que nous l’appréhendons cliniquement [26].
Il est aujourd’hui admis qu’un stress mécanique axonal au niveau de la lame criblée initierait la dégénérescence des CGR. L’hypertension intra-oculaire et, en miroir, les propriétés biomécaniques tissulaires de cette zone favoriseraient ainsi ce stress originel. Cette contrainte s’appliquerait non seulement sur les axones mais aussi sur le circuit vasculaire, créant des conditions d’ischémie locale et fragilisant encore les neurones. Récemment, le rôle de la glie et plus spécifiquement celui de la population astrocytaire spécifique de la tête du nerf optique a été proposé comme un mécanisme essentiel aux altérations axonales au niveau de la lame criblée. Ainsi, les caractéristiques intrinsèques de ces astrocytes associées aux contraintes mécaniques et hyperbares subies semblent à l’origine d’une activation gliale contribuant non seulement à l’augmentation des contraintes mécaniques – hypertrophie cellulaire, remaniement de la matrice extracellulaire – mais aussi à la pathogenèse d’un environnement cellulaire hostile – défaut d’élimination du glutamate extracellulaire, dérégulation de l’homéostasie ionique et en particulier calcique, vasoconstriction et synthèse de médiateurs inflammatoires neurotoxiques.
Comme détaillé précédemment, la capacité neuronale de compartimentalisation de la réaction au stress entraînerait une dégénérescence axonale bien avant la mort somatique des CGR. Ainsi, les contraintes mécaniques et biologiques subies seraient le facteur déclenchant d’une autodestruction axonale, différée, progressive et probablement rétrograde, qui conduirait à une dysfonction du transport synaptique et axonal, puis à une destruction de l’axone.
Les altérations axonales pathologiques seraient à l’origine d’un blocage du transport axonal antérograde et rétrograde des neurotrophines. Cette carence en neurotrophines, ainsi que les altérations des communications synaptiques au niveau central, déclencheraient alors l’apoptose des CGR. En complément, l’activation gliale dérégulerait les apports en facteurs trophiques d’origine gliale et déclencherait aussi l’apoptose neuronale.
La mort initiale de certaines populations de CGR serait à l’origine d’un excès de glutamate de calcium et d’ATP dans le voisinage de celle-ci, conduisant à une excitotoxicité dans le micro-environnement cellulaire proche. Ainsi, la stimulation pathologique des récepteurs au glutamate, NMDA et non NMDA, induirait l’apoptose des CGR environnantes. En parallèle, la dysfonction gliale, en particulier des cellules de Müller, participerait au défaut d’élimination du glutamate, contribuant ainsi au phénomène excitotoxique.
Parallèlement à l’activation gliale initiale liée aux stress ischémiques et hyperbares, la raréfaction progressive des CGR et donc le défaut d’interaction entre neurones et macroglie serait à l’origine d’un remodelage cellulaire et matriciel de type cicatriciel fragilisant l’environnement des cellules rétiniennes encore présentes. De moins en moins stimulées par les neurones, astrocytes et cellules gliales de Müller favoriseraient alors la genèse d’un tissu connectif cicatriciel dont l’environnement ionique et métabolique serait hostile à la fonction et la survie neuronale.
La séquence proposée ci-dessus offre une vue d’ensemble de la chronologie du glaucome, bien que la linéarité temporelle des événements évoqués puisse paraître un peu simpliste. La figure 8-4 tente une synthèse spatiale et temporelle de l’ensemble de nos connaissances actuelles sur la pathogenèse biocellulaire de la neuropathie glaucomateuse. « Le simple est toujours faux. Ce qui ne l’est pas est inutile. »

Fig. 8-4 Modélisation des grands mécanismes pathogéniques impliqués dans la dégénérescence glaucomateuse des CGR. Les chiffres présentés à côté des principaux processus représentent une suggestion chronologique, bien que l’ensemble de ces éléments puissent coexister et/ou survenir dans un ordre différent.
Retenir
La neurodégénérescence rétinienne glaucomateuse résulte de processus pathogéniques complexes et mal compris.
L’apoptose des CGR constitue l’élément central de la neuropathie glaucomateuse, mais d’autres phénomènes de dégénérescence axonale ou synaptique y contribuent en l’absence de tout processus apoptotique initial.
Les relations cellulaires entre neurones et glie semblent aujourd’hui jouer un rôle déterminant dans l’équilibre structural et fonctionnel du tissu rétinien.
Ainsi, une approche intégrée de l’ensemble des interactions et mécanismes qui sous-tendent cette dégénérescence pourrait un jour offrir aux stratégies thérapeutiques de neuroprotection le succès qu’elles ne rencontrent pas aujourd’hui.
[1] Agarwal R, Gupta SK, Agarwal P, et al. Current concepts in the pathophysiology of glaucoma. Ind J Ophthalmol. 2009 ; 57 : 257-66.
[2] AGIS. The Advanced Glaucoma Intervention Study. 7. The relationship between control of intraocular pressure and visual field deterioration. Am J Ophthalmol. 2000 ; 130 : 429-40.
[3] Almasieh M, Wilson AM, Morquette B, et al. The molecular basis of retinal ganglion cell death in glaucoma. Prog Ret Eye Res. 2012 ; 31 : 152-81.
[4] Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974 ; 13 : 771-83.
[5] Bai Y, Shi Z, Zhuo Y, et al. In glaucoma the upregulated truncated TrkC.T1 receptor isoform in glia causes increased TNF-alpha production leading to retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2010 ; 51 : 6639-51.
[6] Baltan S, Inman D, Danilov C, et al. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J Neurosci. 2010 ; 30 : 5644-52.
[7] Baltmr A, Duggan J, Nizari S, et al. Neuroprotection in glaucoma. Is there a future role ? Exp Eye Res. 2010 ; 91 : 554-66.
[8] Band L, Hall C, Richardson G, et al. Intracellular flow in optic nerve axons : a mechanism for cell death in glaucoma. Invest Ophthalmol Vis Sci. 2009 ; 50 : 3750-8.
[9] Bringmann A, Pannicke T, Grosche J, et al. Müller cells in the healthy and diseased retina. Prog Ret Eye Res. 2006 ; 25 : 397-424.
[10] Bull ND, Limb GA, Martin KR. Human Müller stem cell (MIO-M1) transplantation in a rat model of glaucoma : survival, differentiation, and integration. Invest Ophthalmol Vis Sci. 2008 ; 49 : 3449-56.
[11] Chauhan BC, LeVatte TL, Jollimore CA, et al. Model of endothelin-1-induced chronic optic neuropathy in rat. Invest Ophthalmol Vis Sci. 2004 ; 45 : 144-52.
[12] Chen H, Weber AJ. BDNF enhances retinal ganglion cell survival in cats with optic nerve damage. Invest Ophthalmol Vis Sci. 2001 ; 42 : 966-74.
[13] Dreyer EB, Zurakowski D, Schumer RA, et al. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol. 1996 ; 114 : 299-305.
[14] Gregory MS, Hackett CG, Abernathy EF, et al. Opposing roles for membrane bound and soluble Fas ligand in glaucoma-associated retinal ganglion cell death. PLoS One 2011 ; 6 : e17659.
[15] Huang W, Fileta JB, Dobberfuhl A, et al. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci USA. 2005 ; 102 : 12242-7.
[16] Izzotti A, Saccà SC, Cartiglia C, De Flora S. Oxidative deoxyribonucleic acid damage in the eyes of glaucoma patients. Am J Med. 2003 ; 114 : 638-46.
[17] Johnson EC, Jia L, Cepurna WO, et al. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2007 ; 48 : 3161-77.
[18] Kanamori A, Nakamura M, Nakanishi Y, et al. Long-term glial reactivity in rat retinas ipsilateral and contralateral to experimental glaucoma. Exp Eye Res. 2005 ; 81 : 48-56.
[19] Kerrigan LA, Zack DJ, Quigley HA, et al. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol. 1997 ; 115 : 1031-35.
[20] Lambiase A, Aloe L, Centofanti M, et al. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops : implications for glaucoma. Proc Natl Acad Sci USA. 2009 ; 106 : 13469-74.
[21] Leaver SG, Cui Q, Plant GW, et al. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther. 2006 ; 13 : 1328-41.
[22] Lebrun-Julien F, Duplan L, Pernet V, et al. Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J Neurosci. 2009 ; 29 : 5536-45.
[23] Levin LA, Danesh-Meyer HV. Lost in translation : bumps in the road between bench and bedside. JAMA. 2010 ; 303 : 1533-34.
[24] Lin HJ, Tsai FJ, Chen WC, et al. Association of tumour necrosis factor alpha-308 gene polymorphism with primary open-angle glaucoma in Chinese. Eye. 2003 ; 17 : 31-4.
[25] Nakajima Y, Inokuchi Y, Nishi M, et al. Coenzyme Q10 protects retinal cells against oxidative stress in vitro and in vivo. Brain Res. 2008 ; 1226 : 226-33.
[26] Nickells RW. From ocular hypertension to ganglion cell death : a theoretical sequence of events leading to glaucoma. Can J Ophthalmol. 2007 ; 42 : 278-87.
[27] Nickells RW, Semaan SJ, Schlamp CL. Involvement of the Bcl2 gene family in the signaling and control of retinal ganglion cell death. Prog Brain Res. 2008 ; 173 : 423-35.
[28] Osborne NN. Mitochondria : their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res. 2010 ; 90 : 750-7.
[29] Osborne NN. Recent clinical findings with memantine should not mean that the idea of neuroprotection in glaucoma is abandoned. Acta Ophthalmol. 2008 ; 87 : 450-4.
[30] Osborne NN, Wood JP, Chidlow G, et al. Effectiveness of levobetaxolol and timolol at blunting retinal ischaemia is related to their calcium and sodium blocking activities : relevance to glaucoma. Brain Res Bull. 2004 ; 62 : 525-8.
[31] Quigley HA. Neuronal death in glaucoma. Prog Ret Eye Res. 1998 ; 18 : 39-57.
[32] Quigley HA, McKinnon SJ, Zack DJ, et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000 ; 41 : 3460-6.
[33] Ren L, Danias J. A role for complement in glaucoma ? Adv Exp Med Biol. 2010 ; 703 : 95-104.
[34] Sawada H, Fukuchi T, Tanaka T, Abe H. Tumor necrosis factor-a concentrations in the aqueous humor of patients with glaucoma. Invest Ophthalmol Vis Sci. 2010 ; 51 : 903-6.
[35] Schmalbruch H, Jensen HJ, Bjaerg M, et al. A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol. 1991 ; 50 : 192-204.
[36] Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007 ; 131 : 1164-78.
[37] Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci. 2003 ; 44 : 3025-33.
[38] Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. I. Invest Ophthalmol Vis Sci. 1998 ; 39 : 2277-87.
[39] Weber AJ, Chen H, Hubbard WC, Kaufman PL. Experimental glaucoma and cell size, density, and number in the primate lateral geniculate nucleus. Invest Ophthalmol Vis Sci. 2000 ; 41 : 1370-9.
[40] Weber AJ, Harman CD. Structure-function relations of parasol cells in the normal and glaucomatous primate retina. Invest Ophthalmol Vis Sci. 2005 ; 46 : 3197-207.
[41] Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci. 1998 ; 39 : 2304-20.
[42] Whitmore AV, Libby RT, John SWM. Glaucoma : thinking in new ways – a role for autonomous axonal self-destruction and other compartimentalised processes. Prog Ret Eye Res. 2005 ; 24 : 639-62.
[43] Wood JP, Schmidt KG, Melena J, et al. The beta-adrenoceptor antagonists metipranolol and timolol are retinal neuroprotectants : comparison with betaxolol. Exp Eye Res. 2003 ; 76 : 505-16.
[44] Wu Q, Zhang M, Song BW, et al. Expression of ciliary neurotrophic factor after induction of ocular hypertension in the retina of rats. Chin Med J. 2007 ; 120 : 1825-9.
[45] Yuan L, Neufeld AH. Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res. 2001 ; 64 : 523-32.
[46] Zhang X, Li A, Ge J, et al. Acute increase of intraocular pressure releases ATP into the anterior chamber. Exp Eye Res. 2007 ; 85 : 637-43.
[47] Zhong YS, Leung CK, Pang CP. Glial cells and glaucomatous neuropathy. Chin Med J. 2007 ; 20 : 326-35.
A. Labbé
Les mécanismes précis à l’origine de la neuropathie optique glaucomateuse et le rôle exact de la pression intra-oculaire (PIO) demeurent controversés.
La portion laminaire de la tête du nerf optique (TNO) ou lame criblée serait le site principal de lésion des axones des cellules ganglionnaires rétiniennes (CGR) dans le glaucome.
La théorie biomécanique du glaucome intègre les forces et déformations liées à la PIO comme un élément central des modifications physiologiques et pathologiques des tissus de la TNO et de leur vascularisation.
Ces changements tissulaires comprennent non seulement les modifications des tissus conjonctifs de la lame criblée et de la sclère péripapillaire, mais aussi des composants cellulaires de ces tissus, des axones des CGR et de leur vascularisation.
Le glaucome primitif à angle ouvert (GPAO) est une neuropathie optique progressive liée à la perte des cellules ganglionnaires rétiniennes et associant une atteinte caractéristique de la papille et du champ visuel. Alors que les changements cliniques au niveau de la papille ont été largement décrits, les mécanismes précis à l’origine de cette neuropathie demeurent controversés [8, 10, 14, 41]. Les théories qui ont tenté d’expliquer la physiopathogénie des glaucomes ont été le plus souvent regroupées en deux catégories : mécaniques, liées à la PIO, et vasculaires, liées au flux sanguin dans la TNO [14]. Une hypothèse plus récente serait la combinaison de ces deux mécanismes au sein d’une théorie biomécanique de la neuropathie glaucomateuse [8, 10]. L’absence de description d’un mécanisme précis à l’origine des changements cliniques et tissulaires observés dans le glaucome pourrait être due au fait qu’il n’existe en réalité pas un mécanisme unique, mais plusieurs mécanismes intriqués de manière complexe.
À l’image de la physiopathogénie du glaucome, jusqu’à sa définition même, le rôle exact de la PIO dans le GPAO est aussi très largement discuté [8]. En effet, alors que certains patients ayant une PIO dite normale développent un glaucome, d’autres ayant une PIO élevée ne présentent pas les signes de la maladie. Néanmoins, abaisser la PIO est le seul traitement qui ait prouvé son efficacité pour retarder l’apparition ou ralentir la progression du glaucome. Les relations entre la PIO et le glaucome, c’est-à-dire les phénomènes biomécaniques impliqués dans cette neuropathie optique, demeurent au centre du développement et de la progression de cette pathologie.
En termes biomécaniques, la papille ou TNO représente une zone particulière car elle constitue un point faible dans l’enveloppe cornéosclérale résistante du globe oculaire. De nombreux éléments suggèrent ainsi que la portion laminaire de la TNO ou lame criblée soit le site principal de lésion des axones des CGR dans le glaucome [27, 34, 35, 39, 41].
La papille ou TNO peut être divisée au niveau histologique en quatre régions, de la plus superficielle à la plus profonde : la couche des fibres nerveuses rétiniennes, la portion prélaminaire, la portion laminaire ou lame criblée, et la portion rétrolaminaire. La lame criblée correspond à un tamis à plusieurs étages constitué de feuillets de tissus conjonctifs fenêtrés. Ces feuillets cribriformes sont composés d’un centre de matrice extracellulaire (MEC) fait de collagène et de fibres élastiques, de capillaires entourés par une membrane basale et de quelques cellules d’aspect fibroblastique appelées cellules de la lame criblée. Des astrocytes recouvrent ces feuillets et envoient des prolongements entre les axones des cellules ganglionnaires. Ces cellules gliales produisent également une membrane basale continue qui les sépare des feuillets cribriformes. Ces lames de tissu conjonctif sont perforées par 200 à 400 orifices irréguliers de tailles variables formant des canaux à travers lesquels passent les axones des CGR.
La fonction principale de la lame criblée est de permettre le passage des axones des CGR et des vaisseaux centraux de la rétine au travers du canal scléral tout en préservant ces structures du gradient de pression entre les espaces intra- et extra-oculaires. La lame criblée offre ainsi un support structurel mais aussi fonctionnel pour l’ensemble des axones des CGR qui la traverse ; elle serait également le principal site d’atteinte de ces dernières dans la neuropathie optique glaucomateuse [27, 34, 35, 39, 41].
Les travaux de Quigley et al. [34, 39] ont permis de démontrer que l’excavation glaucomateuse résultait de l’étirement, de la compression et du réarrangement des tissus conjonctifs au sein de la tête du nerf optique, en particulier de la lame criblée, en réponse à l’élévation de la PIO. De nombreux changements apparaissent au niveau de la lame criblée en cas d’élévation de la PIO [12, 39]. Les feuillets cribriformes de la lame criblée s’effondrent comme un accordéon qui se ferme sous l’effet d’une compression mécanique verticale, faisant reculer la base de l’excavation et refoulant la lame criblée vers l’extérieur (fig. 8-5 et fig. 8-6) [34]. Cette compression des feuillets de la lame criblée entraînerait également une compression des axones des CGR et serait ainsi la cause de la neuropathie optique glaucomateuse. Des études en microscopie électronique sur des glaucomes expérimentaux ont ainsi confirmé que les axones des CGR étaient lésés lorsque la lame criblée était distendue et collabée [36]. Il semblerait que même un déplacement mineur des orifices de la lame criblée ait un retentissement sur le fonctionnement des axones des CGR [28]. L’analyse des mouvements des feuillets cribriformes dans le glaucome, avec un déplacement postérieur et une rotation autour de leur ancrage scléral, a également montré qu’ils induisaient les forces de compression les plus importantes sur les axones de la périphérie du nerf optique (fig. 8-7), correspondant aux déficits du champ visuel observés en pratique clinique dans le glaucome [38, 36, 42]. L’augmentation de la taille de l’excavation résulte ainsi de la perte des fibres nerveuses rétiniennes et du réarrangement tissulaire lié à la compression des feuillets cribriformes de la lame criblée [39]. Les pores plus larges et le manque relatif de tissu de soutien dans les zones inférieures et supérieures de la lame criblée pourraient expliquer les lésions plus importantes observées dans ces régions par rapport aux régions nasales et temporales [36, 43].

Fig. 8-5 Image en microscopie électronique après digestion à la trypsine de la TNO d’un sujet normal (a) et d’un patient avec un glaucome avancé (b). Chez le patient glaucomateux, on observe une excavation postérieure en arrière du canal scléral et une rotation des feuillets de la lame criblée. (Source : Crawford Downs et al., 2011 [10]. Reproduction autorisée.)

Fig. 8-6 Image en tomographie par cohérence optique de la papille et de la lame criblée chez un sujet normal (a) et chez un sujet glaucomateux (b). Chez le patient glaucomateux, la lame criblée est plus fine et présente un bombement postérieur très important. (Collection Y. Wang, Beijing Institute of Ophthalmology, Pékin, Chine.)

Fig. 8-7 Photographie de la papille d’une patiente de 68 ans qui présente un glaucome chronique à angle ouvert (a). Image en optique adaptative de la même papille montrant un élargissement des pores de la lame criblée (b) (RTX1, Imagine Eyes, Orsay, France). (Collection S. Carette-Zwillinger.)
Le glaucome chronique s’accompagne d’un remodelage tissulaire de la lame criblée. Les composants de la MEC de la lame criblée ont donc un rôle important dans les changements de la TNO induits par le glaucome.
Dans les feuillets cribriformes de la lame criblée, des masses granulaires d’élastine apparaissent et les fibres d’élastine sont progressivement désorganisées avec la progression de la maladie [17]. Certains auteurs ont décrit une augmentation de la quantité d’élastine au sein de la lame criblée des yeux glaucomateux par rapport à des sujets contrôles du même âge [32]. Parallèlement, il pourrait également exister une perte d’élastine au niveau des feuillets cribriformes situés à la périphérie du nerf optique dans les glaucomes sévères [17]. Des analyses ultrastructurales ont révélé des anomalies dans la morphologie des fibres élastiques de la lame criblée avec une fragmentation et une accumulation de matériel non fibrillaire de type élastique en amas au sein de la lame criblée ou dans la région de l’insertion dans les yeux glaucomateux [20]. Ces modifications s’accompagnent d’une diminution de la densité des fibres de collagène au niveau du corps des feuillets de la lame criblée [20, 38]. Les fibres de collagène et élastiques auraient un rôle complémentaire au sein du tissu conjonctif de la lame criblée pour assurer la protection mécanique de cette structure. L’élastine plus élastique réagirait pour des niveaux faibles de distorsion et les fibres solides de collagène pour des niveaux plus importants. Les modifications de ces deux composants tissulaires altéreraient donc significativement les propriétés mécaniques du tissu lors de la maladie initiale, compromettant les capacités de la lame criblée à supporter le stress engendré par l’élévation ou les fluctuations de la PIO [18]. Ces changements participent très certainement aussi à l’effondrement et à la compression de la lame criblée à des stades plus avancés [18, 39]. Le collagène de type VI, que l’on retrouve dans de nombreux tissus en réponse à un stress tissulaire, augmente également au cœur des feuillets de la lame criblée dans le glaucome [18]. Enfin, la densité de la MEC s’accroît au niveau de la membrane basale dans les régions laminaires et prélaminaires des yeux glaucomateux [17]. Ces observations ont été confirmées sur des modèles expérimentaux de glaucome chez le primate et retrouvent un volume de tissu conjonctif de la lame criblée 80 % plus important dans les yeux glaucomateux par rapport à l’œil controlatéral indemne de glaucome [47].
Ces changements tissulaires importants au niveau de la lame criblée suggèrent à la fois une synthèse et une dégradation anormale de la MEC dans le glaucome résultant d’une réponse cellulaire aux modifications du micro-environnement de la TNO [10, 18].
En plus du tissu glial et des axones, un réseau vasculaire est naturellement présent au niveau de la TNO et de la lame criblée (voir chapitre 6). La distribution et l’organisation particulière de ce réseau pourraient entraîner une susceptibilité accrue aux atteintes ischémiques, notamment dans les régions temporales supérieures et inférieures, expliquant ainsi les atteintes du champ visuel observées dans le glaucome [16]. Ces éléments et le rôle du système microvasculaire dans le développement de la neuropathie optique glaucomateuse sont abordés en détail dans le chapitre 8-III.
Afin d’intégrer les différents acteurs tissulaires et cellulaires impliqués à la physiopathogénie de la neuropathie optique glaucomateuse, certains auteurs considèrent la TNO comme une structure biomécanique [8, 10, 11]. Ainsi, les forces et déformations liées à la PIO seraient un déterminant central des modifications physiologiques mais aussi pathologiques des tissus de la TNO et de leur vascularisation. Ces changements comprennent non seulement les modifications des tissus conjonctifs de la lame criblée et de la sclère péripapillaire précédemment décrits, mais aussi des composants cellulaires de ces tissus et des axones des CGR ainsi que de leur vascularisation (fig. 8-8).
La biomécanique d’un tissu inclut à la fois la réponse aiguë d’un tissu à une force mécanique mais également les changements morphologiques et microstructuraux induits à long terme sur cette même structure [8, 10]. Selon la théorie biomécanique de la neuropathie optique glaucomateuse, la susceptibilité du nerf optique à l’élévation de la PIO est fonction de ces deux types de réponse, aiguë et chronique [8]. Des yeux avec une combinaison particulière dans l’organisation mais aussi dans la composition des tissus de la TNO seraient ainsi plus sujets à une modification de la PIO [10]. De nombreux auteurs ont émis l’hypothèse que les déformations induites par la PIO entraîneraient une atteinte aiguë des pores de la lame criblée qui aboutirait finalement à l’excavation du nerf optique [8, 40, 44, 46, 48]. Néanmoins, aucune étude n’a montré que des lésions aiguës au niveau de la lame criblée pouvaient être responsables d’une excavation de la TNO d’aspect glaucomateux [10]. La déformation du tissu conjonctif et l’excavation typiquement observée chez l’homme et dans les glaucomes expérimentaux seraient donc plutôt liées à un phénomène chronique.
La relation entre la PIO et la perte des axones de cellules ganglionnaires est extrêmement complexe. Néanmoins, le niveau de la PIO demeure un élément essentiel de la survie des axones des CGR. Ainsi, même de petites différences en termes de PIO entre les deux yeux d’un même patient sont corrélées à des différences en termes d’évolution du glaucome [9]. De la même manière, les variations de la PIO pourraient également avoir une influence, les patients subissant les plus grandes variations de PIO ayant le plus de risque de progression de leur glaucome [3, 31]. Cela correspond bien à l’idée que des forces et des déformations appliquées à la lame criblée à travers des éléments cellulaires pourraient être responsables de la perte des axones des cellules ganglionnaires. Les cellules gliales non neuronales joueraient ainsi un rôle majeur dans l’intégration des effets de l’élévation et des fluctuations de la PIO en association avec les modifications vasculaires induites [8, 10]. Pour chaque patient, l’importance relative de chacun des éléments demeure extrêmement variable.
Les astrocytes sont les cellules gliales prédominantes au niveau de la TNO [19]. Ils entourent les orifices de la lame criblée et communiquent par l’intermédiaire de jonctions cellulaires pour former un réseau protecteur pour les axones des CGR (voir chapitre 6) [19]. Ils ont un rôle à la fois fonctionnel dans le maintien d’un environnement extracellulaire permettant la conduction du potentiel d’action des axones des CGR, mais aussi structurel : les astrocytes fabriquent la membrane basale et sont impliqués dans la production de collagène et d’élastine de la lame criblée [19]. Leur position entre la lame criblée et les axones des CGR leur confèrent donc un rôle essentiel pour transférer aux axones les modifications de la lame criblée. De même, si les capacités des astrocytes sont compromises, cela va entraîner une atteinte des axones des cellules ganglionnaires [10]. D’autres cellules gliales ont été identifiées au sein des feuillets de la lame criblée [25, 26]. Ces cellules de la lame criblée pourraient également intervenir dans le remodelage de la lame criblée dans le glaucome.
Les astrocytes et les cellules de la lame criblée jouent certainement un rôle central dans le remodelage de la lame criblée et les atteintes des fibres nerveuses liées à l’élévation de la PIO dans le glaucome [19, 29]. Plusieurs études ont observé le comportement biomécanique de ces cellules en réponse à une élévation de la PIO [23, 24, 52]. Des cultures de cellules de la lame criblée ont ainsi montré une sécrétion augmentée de collagène de type I sous l’effet de l’élévation de la pression hydrostatique [52]. Lorsque ces cellules sont soumises à une pression ou à des déformations, elles sur-expriment également le TGF-β2 (transforming growth factor-β2), facteur important de la régulation du remodelage de la MEC [24]. Les métalloprotéinases (MMP) ont un rôle central dans les processus de remodelage de la MEC. Une augmentation de la production de MMP-2 a ainsi été observée lorsque les cellules de la lame criblée étaient cultivées dans un environnement soumis à un stress mécanique répété [23]. De même, une sur-expression de MMP a été retrouvée dans la lame criblée des primates avec un glaucome expérimental [1]. D’autres protéines comme les intégrines, qui recouvrent la membrane basale laminaire des astrocytes et accrochent le cytosquelette des cellules à la MEC environnante, pourraient également être un lien important entre la déformation de la lame criblée, les forces appliquées sur les cellules liées à la PIO et la réponse cellulaire en termes de remodelage de la MEC mais aussi d’atteinte des axones des CGR [30].
L’ensemble de ces résultats supporte l’hypothèse qu’une élévation de la PIO et une atteinte des cellules de la lame criblée ou une diminution du flux sanguin dans cette région entraîneraient le remodelage de la MEC observé dans le glaucome. De manière intéressante, ces mécanismes potentiels sont tous deux liés à une élévation chronique de la PIO et ne sont pas simplement l’effet d’une atteinte axonale ou de la mort de ces axones [10].

Fig. 8-8 Théorie biomécanique de la neuropathie optique glaucomateuse. La PIO génère des forces et entraîne des déformations des tissus de la tête du nerf optique. Ces déformations dépendent de la géométrie et des propriétés mécaniques individuelles de ces tissus. Les forces induites par la PIO peuvent modifier le flux sanguin dans la région laminaire de la TNO et également entraîner des altérations directes et indirectes des tissus conjonctifs. Enfin, ces changements tissulaires altèrent à leur tour les propriétés biomécaniques de la TNO et modifient ainsi la réponse de ces tissus aux forces et déformations induites par la PIO. (D’après Crawford Downs et al., 2011 [10].)
Au niveau des axones des CGR, le transport axonal rétrograde et orthograde serait bloqué au niveau des orifices de la lame criblée dans les modèles expérimentaux de glaucome [27, 33, 37]. Cependant, même si les changements morphologiques de la lame criblée semblent être un des éléments centraux dans l’atteinte des axones des CGR lors d’un stade avancé de glaucome, aux stades initiaux du glaucome, la lame criblée ne paraît pas encore modifiée [28]. Les changements structurels au niveau de la lame criblée seraient ainsi précédés par des changements au niveau du transport axonal. Grâce aux modèles biomécaniques de la TNO [7, 8], il semblerait qu’il puisse exister un stress sur les axones des CGR au niveau de la lame criblée rapidement après une élévation de la PIO [7]. Les forces qui s’exercent sur le nerf optique sont dynamiques et le degré avec lequel la lame criblée peut se déformer varie avec l’âge : très élastique chez le sujet jeune ; diminution de l’élasticité chez le sujet âgé [2]. Des études récentes ont ainsi montré que la lame criblée augmentait d’épaisseur après une élévation de la PIO de courte durée chez le singe, confirmant le caractère dynamique des changements au niveau de la TNO [51].
Des études sur l’effet de l’élévation de la PIO sur l’anatomie des axones ont révélé un gonflement des axones au niveau du site de compression dans les feuillets de la lame criblée [28, 35]. Les axones sont importants pour réguler l’activité et la survie du corps cellulaire, ce qui pourrait expliquer la mort des CGR dans le glaucome. Par ailleurs, la mort des CGR peut elle-même influencer la survie des cellules ganglionnaires voisines, notamment par la libération de neuromédiateurs [28]. Les mécanismes qui concourent à la perte des CGR dans le glaucome restent néanmoins très discutés.
L’ élévation et les fluctuations de la PIO semblent avoir un rôle central dans la physiopathogénie du GPAO ; néanmoins, l’existence de glaucomes à PIO normale semble remettre en question la théorie biomécanique du développement de cette neuropathie optique. Des études ont démontré une association entre le glaucome à pression normale et l’hypotension artérielle nocturne et des phénomènes vasospastiques. Il a donc été émis l’hypothèse que les facteurs vasculaires pourraient jouer un rôle primordial dans la pathogénie du glaucome à pression normale [6, 13, 15, 22]. Toutefois, la morphologie de la TNO dans les glaucomes à pression normale, semblable à celle observée dans les glaucomes à pression élevée mais différente de celle objectivée dans les atteintes vasculaires de la TNO, n’est pas en accord avec cette hypothèse [22].
Selon certains auteurs et notamment Jonas [22], l’élément central dans la physiopathogénie du glaucome pourrait être non pas la PIO (ou pression transcornéenne), mais le gradient de pression au niveau de la lame criblée. Ce dernier dépend de la différence de pression et de la distance entre le compartiment intra-oculaire et le compartiment rétrobulbaire. La distance entre ces deux compartiments dépend essentiellement de l’épaisseur de la lame criblée. Ainsi, l’amincissement de la lame criblée des yeux fortement myopes pourrait expliquer leur plus grande susceptibilité au glaucome [21, 22, 50]. Également, ce même amincissement observé dans les stades avancés de glaucome pourrait expliquer pourquoi la progression du glaucome est plus rapide à ce stade [22]. La différence de pression au travers de la lame criblée (translaminaire) dépend quant à elle de la PIO et de la pression du liquide céphalorachidien (LCR) rétrobulbaire. Plusieurs auteurs ont ainsi observé qu’une pression faible du LCR pouvait être associée à une neuropathie optique glaucomateuse [4, 5, 22, 49]. Cela a par ailleurs été confirmé sur des modèles animaux expérimentaux [22]. Dans une vaste étude rétrospective reprenant les cas de patients ayant eu une ponction lombaire avec mesure de la pression du LCR, il a ainsi été observé une pression du LCR plus faible dans le groupe des patients glaucomateux par rapport aux sujets indemnes de glaucome [4]. Cela a également été retrouvé pour le glaucome à pression normale [5]. De manière intéressante, dans une autre étude, la baisse de pression du LCR a été statistiquement corrélée avec la perte du champ visuel, ce qui n’était pas le cas de la PIO seule dans cette étude [22, 45].
La baisse nocturne de la pression artérielle s’associe physiologiquement à une baisse de la pression du LCR. Celle-ci pourrait entraîner ainsi une augmentation de la pression au travers de la lame criblée et, par voie de conséquence, des modifications identiques à ce qui est observé en cas d’élévation de la PIO [22]. Outre la physiopathologie du glaucome à pression normale, ce modèle expliquerait aussi l’association entre le glaucome à pression normale et une pression artérielle systémique abaissée [22].
L’analyse du rôle de la pression du LCR dans le développement de la neuropathie optique glaucomateuse est cependant limitée par la difficulté et le caractère invasif de cette mesure en pratique. Néanmoins, il semblerait que l’imagerie par résonance magnétique des espaces sous-arachnoïdiens entourant le nerf optique puisse être un marqueur indirect non invasif corrélé à la pression du LCR. Une étude très récente a ainsi montré un rétrécissement des espaces sous-arachnoïdiens entourant le nerf optique, suggérant une diminution de la pression du LCR chez les patients avec un glaucome à pression normale par rapport aux patients qui présentaient un glaucome à pression élevée (voir fig. 8-8) [49].
La physiopathogénie du glaucome comme le rôle exact de la PIO demeurent très controversés. Néanmoins, il semblerait qu’une approche biomécanique des modifications tissulaires et cellulaires au niveau de la lame criblée puisse expliquer l’ensemble des éléments cliniques mais aussi histologiques observés chez les patients glaucomateux. Ces études suggèrent que le remodelage de la matrice extracellulaire serait lié à des forces appliquées à la TNO qui dépassent la tolérance physiologique des cellules présentes. Cela suggère une réponse des cellules à des modifications biomécaniques qui modifient à leur tour leur environnement pour revenir à une homéostasie de leur environnement mécanique [10]. Ainsi, au stade débutant du glaucome, une modification du rôle protecteur du tissu conjonctif et de ses acteurs cellulaires, associée à une atteinte progressive des axones, pourraient être les deux mécanismes principaux en réponse à l’élévation de la PIO. Lors d’un stade plus tardif, la déformation importante de la lame criblée associée à un remodelage de la MEC du tissu conjonctif en lien avec les acteurs cellulaires pourrait expliquer les lésions observées.
La compréhension de la physiopathologie du glaucome reste encore limitée par l’absence d’analyse in vivo chez l’homme des modifications tissulaires liées à l’élévation de la PIO. De nouvelles techniques d’imagerie mais aussi de nouveaux modèles numériques permettront certainement de mieux comprendre les mécanismes impliqués dans le futur.
Retenir
L’ensemble des mécanismes physiopathogéniques du GPAO n’est pas encore totalement élucidé.
Les modifications tissulaires et cellulaires biomécaniques au niveau de la lame criblée pourraient expliquer les altérations histologiques à l’origine des manifestations cliniques de la neuropathie optique.
[1] Agapova OA, Kaufman PL, Lucarelli MJ, et al. Differential expression of matrix metalloproteinases in monkey eyes with experimental glaucoma or optic nerve transection. Brain Res. 2003 ; 967 : 132-43.
[2] Albon J, Purslow PP, Karwatowski WS, Easty DL. Age related compliance of the lamina cribrosa in human eyes. Br J Ophthalmol. 2000 ; 84 : 318-23.
[3] Asrani S, Zeimer R, Wilensky J, et al. Large diurnal fluctuations in intraocular pressure are an independent risk factor in patients with glaucoma. J Glaucoma. 2000 ; 9 : 134-42.
[4] Berdahl JP, Allingham RR, Johnson DH. Cerebrospinal fluid pressure is decreased in primary open-angle glaucoma. Ophthalmology. 2008 ; 115 : 763-8.
[5] Berdahl JP, Fautsch MP, Stinnett SS, Allingham RR. Intracranial pressure in primary open angle glaucoma, normal tension glaucoma, and ocular hypertension : a case-control study. Invest Ophthalmol Vis Sci. 2008 ; 49 : 5412-8.
[6] Broadway DC, Drance SM. Glaucoma and vasospasm. Br J Ophthalmol. 1998 ; 82 : 862-70.
[7] Burgoyne CF, Downs JC, Bellezza AJ, Hart RT. Three-dimensional reconstruction of normal and early glaucoma monkey optic nerve head connective tissues. Invest Ophthalmol Vis Sci. 2004 ; 45 : 4388-99.
[8] Burgoyne CF, Downs JC, Bellezza AJ, et al. The optic nerve head as a biomechanical structure : a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog Retin Eye Res. 2005 ; 24 : 39-73.
[9] Cartwright MJ, Anderson DR. Correlation of asymmetric damage with asymmetric intraocular pressure in normal-tension glaucoma (low-tension glaucoma). Arch Ophthalmol. 1988 ; 106 : 898-900.
[10] Crawford Downs J, Roberts MD, Sigal IA. Glaucomatous cupping of the lamina cribrosa : a review of the evidence for active progressive remodeling as a mechanism. Exp Eye Res. 2011 ; 93 : 133-40.
[11] Downs JC, Roberts MD, Burgoyne CF. Mechanical environment of the optic nerve head in glaucoma. Optom Vis Sci. 2008 ; 85 : 425-35.
[12] Emery JM, Landis D, Paton D, et al. The lamina cribrosa in normal and glaucomatous human eyes. Trans Am Acad Ophthalmol Otolaryngol. 1974 ; 78 : OP290-7.
[13] Emre M, Orgul S, Gugleta K, Flammer J. Ocular blood flow alteration in glaucoma is related to systemic vascular dysregulation. Br J Ophthalmol. 2004 ; 88 : 662-6.
[14] Fechtner RD, Weinreb RN. Mechanisms of optic nerve damage in primary open angle glaucoma. Surv Ophthalmol. 1994 ; 39 : 23-42.
[15] Hayreh SS, Zimmerman MB, Podhajsky P, Alward WL. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994 ; 117 : 603-24.
[16] Hayreh SS. Optic nerve head blood supply in health and disease. In : Lambrou GN, Greve EL (eds). Ocular blood flow in glaucoma : means, methods, and measurements. Amsterdam, Kugler and Ghedini, 1989.
[17] Hernandez MR, Andrzejewska WM, Neufeld AH. Changes in the extracellular matrix of the human optic nerve head in primary open-angle glaucoma. Am J Ophthalmol. 1990 ; 109 : 180-8.
[18] Hernandez MR, Gong, H. Extracellular matrix of the trabecular meschwork and optic nerve head. In : Ritch R, Shields MB, Krupin T (eds). The glaucomas : basic sciences. St Louis, Mosby, 1996 : 213-49.
[19] Hernandez MR. The optic nerve head in glaucoma : role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000 ; 19 : 297-321.
[20] Hernandez MR. Ultrastructural immunocytochemical analysis of elastin in the human lamina cribrosa. Changes in elastic fibers in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1992 ; 33 : 2891-903.
[21] Jonas JB, Berenshtein E, Holbach L. Lamina cribrosa thickness and spatial relationships between intraocular space and cerebrospinal fluid space in highly myopic eyes. Invest Ophthalmol Vis Sci. 2004 ; 45 : 2660-5.
[22] Jonas JB. Role of cerebrospinal fluid pressure in the pathogenesis of glaucoma. Acta Ophthalmol. 2011 ; 89 : 505-14.
[23] Kirwan RP, Crean JK, Fenerty CH, et al. Effect of cyclical mechanical stretch and exogenous transforming growth factor-beta1 on matrix metalloproteinase-2 activity in lamina cribrosa cells from the human optic nerve head. J Glaucoma. 2004 ; 13 : 327-34.
[24] Kirwan RP, Fenerty CH, Crean J, et al. Influence of cyclical mechanical strain on extracellular matrix gene expression in human lamina cribrosa cells in vitro. Mol Vis. 2005 ; 11 : 798-810.
[25] Kirwan RP, Leonard MO, Murphy M, et al. Transforming growth factor-beta-regulated gene transcription and protein expression in human GFAP-negative lamina cribrosa cells. Glia. 2005 ; 52 : 309-24.
[26] Lambert W, Agarwal R, Howe W, et al. Neurotrophin and neurotrophin receptor expression by cells of the human lamina cribrosa. Invest Ophthalmol Vis Sci. 2001 ; 42 : 2315-23.
[27] Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci. 1977 ; 16 : 426-41.
[28] Morgan J. Pathogenesis of glaucomatous optic neuropathy. In : Sharrawy TM, Hitchings R, Crowston J (eds). Glaucoma. Londres, Saunders-Elsevier, 2009 : 45-54.
[29] Morgan JE. Optic nerve head structure in glaucoma : astrocytes as mediators of axonal damage. Eye (Lond). 2000 ; 14 : 437-44.
[30] Morrison JC. Integrins in the optic nerve head : potential roles in glaucomatous optic neuropathy (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc. 2006 ; 104 : 453-77.
[31] Nouri-Mahdavi K, Hoffman D, Coleman AL, et al. Predictive factors for glaucomatous visual field progression in the Advanced Glaucoma Intervention Study. Ophthalmology. 2004 ; 111 : 1627-35.
[32] Pena JD, Netland PA, Vidal I, et al. Elastosis of the lamina cribrosa in glaucomatous optic neuropathy. Exp Eye Res. 1998 ; 67 : 517-24.
[33] Quigley H, Anderson DR. The dynamics and location of axonal transport blockade by acute intraocular pressure elevation in primate optic nerve. Invest Ophthalmol. 1976 ; 15 : 606-16.
[34] Quigley HA, Addicks EM, Green WR, Maumenee AE. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch Ophthalmol. 1981 ; 99 : 635-49.
[35] Quigley HA, Addicks EM. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980 ; 19 : 137-52.
[36] Quigley HA, Addicks EM. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch Ophthalmol. 1981 ; 99 : 137-43.
[37] Quigley HA, Anderson DR. Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Invest Ophthalmol Vis Sci. 1977 ; 16 : 640-4.
[38] Quigley HA, Dorman-Pease ME, Brown AE. Quantitative study of collagen and elastin of the optic nerve head and sclera in human and experimental monkey glaucoma. Curr Eye Res. 1991 ; 10 : 877-88.
[39] Quigley HA, Hohman RM, Addicks EM, et al. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am J Ophthalmol. 1983 ; 95 : 673-91.
[40] Quigley HA. Glaucoma : macrocosm to microcosm the Friedenwald lecture. Invest Ophthalmol Vis Sci. 2005 ; 46 : 2662-70.
[41] Quigley HA. New paradigms in the mechanisms and management of glaucoma. Eye (Lond). 2005 ; 19 : 1241-8.
[42] Radius RL, Anderson DR. The course of axons through the retina and optic nerve head. Arch Ophthalmol. 1979 ; 97 : 1154-8.
[43] Radius RL, Gonzales M. Anatomy of the lamina cribrosa in human eyes. Arch Ophthalmol. 1981 ; 99 : 2159-62.
[44] Radius RL. Anatomy of the optic nerve head and glaucomatous optic neuropathy. Surv Ophthalmol. 1987 ; 32 : 35-44.
[45] Ren R, Jonas JB, Tian G, et al. Cerebrospinal fluid pressure in glaucoma : a prospective study. Ophthalmology. 2010 ; 117 : 259-66.
[46] Ren R, Wang N, Li B, et al. Lamina cribrosa and peripapillary sclera histomorphometry in normal and advanced glaucomatous Chinese eyes with various axial length. Invest Ophthalmol Vis Sci. 2009 ; 50 : 2175-84.
[47] Roberts MD, Grau V, Grimm J, et al. Remodeling of the connective tissue microarchitecture of the lamina cribrosa in early experimental glaucoma. Invest Ophthalmol Vis Sci. 2009 ; 50 : 681-90.
[48] Sigal IA, Ethier CR. Biomechanics of the optic nerve head. Exp Eye Res. 2009 ; 88 : 799-807.
[49] Wang N, Xie X, Yang D, et al. Orbital cerebrospinal fluid space in glaucoma : the Beijing intracranial and intraocular pressure (iCOP) study. Ophthalmology. 2012 ; 119 : 2065-73.
[50] Xu L, Wang Y, Wang S, et al. High myopia and glaucoma susceptibility the Beijing Eye Study. Ophthalmology. 2007 ; 114 : 216-20.
[51] Yang H, Downs JC, Girkin C, et al. 3-D histomorphometry of the normal and early glaucomatous monkey optic nerve head : lamina cribrosa and peripapillary scleral position and thickness. Invest Ophthalmol Vis Sci. 2007 ; 48 : 4597-607.
[52] Yang JL, Neufeld AH, Zorn MB, Hernandez MR. Collagen type I mRNA levels in cultured human lamina cribrosa cells : effects of elevated hydrostatic pressure. Exp Eye Res. 1993 ; 56 : 567-74.
C. Chiquet, B. Mottet, F. Aptel, M. Geiser, J.-P. Romanet
L’atteinte vasculaire associée au glaucome est présente dans les différents lits vasculaires oculaires (de l’artère ophtalmique jusqu’au lit vasculaire rétinien).
Elle se caractérise par une diminution du flux sanguin basal associée à une vasorégulation anormale.
La physiopathologie du glaucome est complexe, impliquant en premier lieu l’hypertension intra-oculaire (HTO) dont le traitement est primordial pour ralentir et limiter l’évolution de la neuropathie optique glaucomateuse. D’autres facteurs pathogéniques ont été plus récemment mis en évidence, comme l’implication de facteurs vasculaires, génétiques, inflammatoires et/ou immunologiques. Des dysrégulations vasculaires au sein du nerf optique sont susceptibles d’aggraver les atteintes histologiques et cliniques de la neuropathie optique glaucomateuse. Les mécanismes impliqués sont liés à une hypoperfusion du nerf optique, intermittente ou chronique, par réduction de la pression de perfusion oculaire et/ou atteinte de l’autorégulation vasculaire. Une réduction de la perfusion oculaire (fig. 8-9) est susceptible d’aggraver le gradient de pression au niveau de la lame criblée provoqué par l’HTO, d’induire une perte de l’autorégulation, des anomalies du couplage neurovasculaire, une ischémie et la cascade du stress oxydant. Les conséquences des dysfonctionnements de la physiologie vasculaire du nerf optique dans le cadre du glaucome dépendent : (i) de la localisation des dysrégulations vasculaires (nerf optique, choroïde, rétine, artère ophtalmique – AO), et (ii) des facteurs biologiques (fig. 8-10) impliqués au niveau de l’œil (oxyde nitrique, endothéline, prostacyclines, mécanismes myogéniques, etc.) ou au niveau du système vasculaire général (pression artérielle, vasospasticité).
La compréhension de la physiopathogénie vasculaire du glaucome nécessite : (i) de bien connaître les techniques de mesure du flux sanguin oculaire (FSO) (pression de perfusion oculaire, flux sanguin ophtalmique, choroïdien, rétinien ou de la tête du nerf optique) qui sont l’objet d’un autre chapitre dans ce rapport, et (ii) de caractériser la topographie et les mécanismes impliqués de l’atteinte vasculaire dans cette neuropathie.
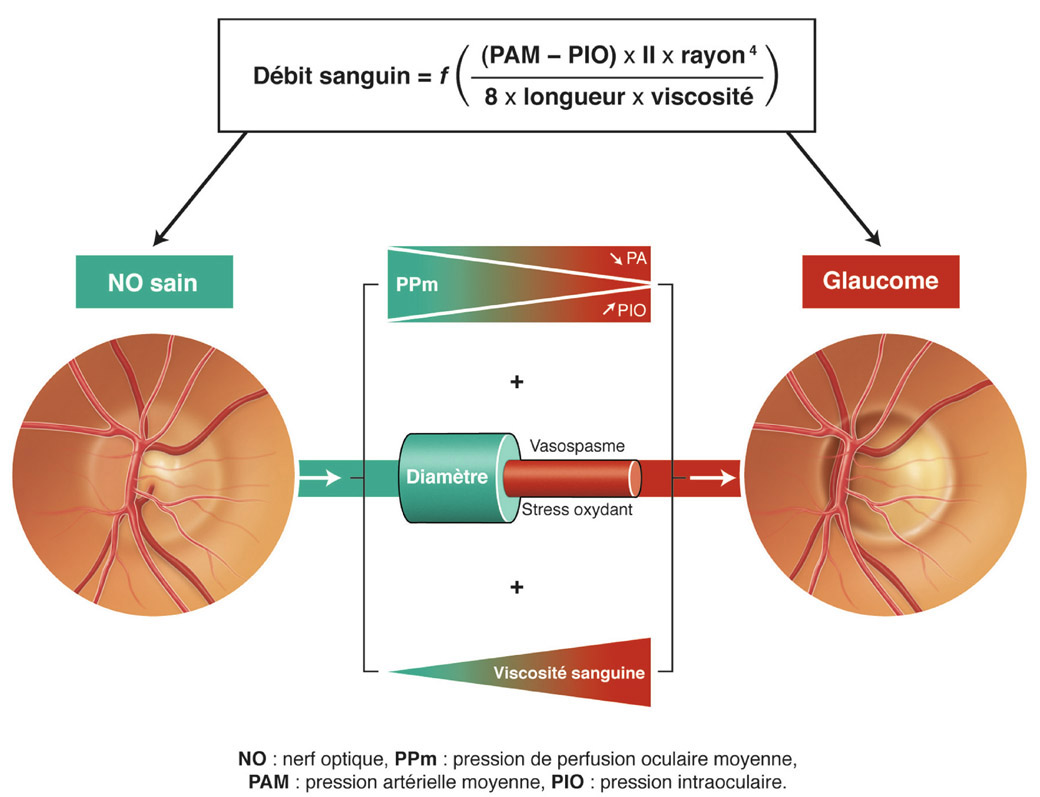
Fig. 8-9 Les facteurs vasculaires du glaucome.
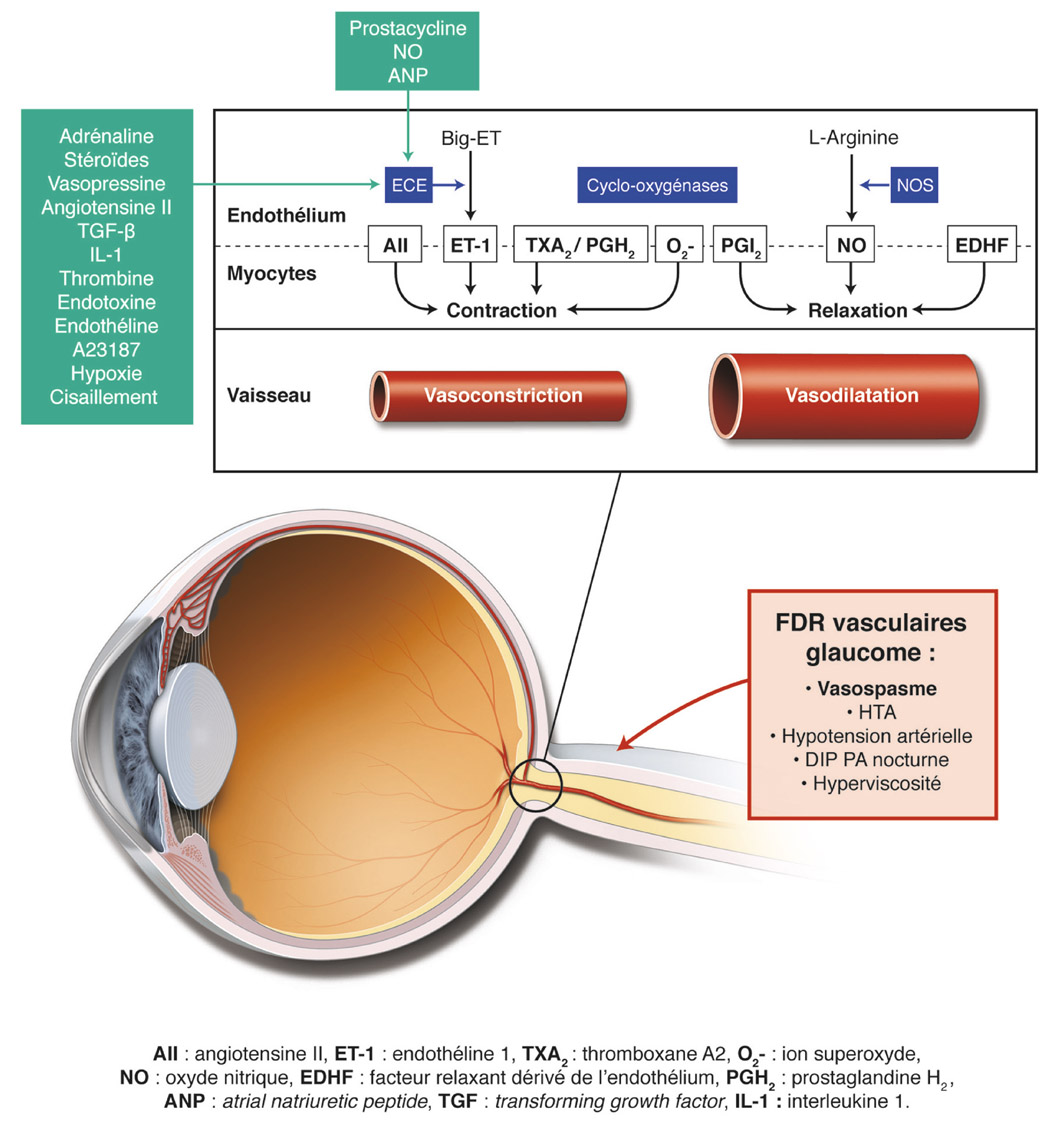
Fig. 8-10 Caractéristiques vasculaires des glaucomes : perte de l’autorégulation et diminution de la PPm.
La majorité des études suggèrent que le FSO est globalement diminué et que la vasoréactivité vasculaire oculaire est réduite chez les patients atteints de glaucome primitif à angle ouvert (GPAO).
De nombreuses études rapportent une réduction de la pression de perfusion oculaire moyenne (PPm) [31, 53], de la pression de perfusion oculaire diastolique (PPd) [1, 24, 30, 54] et/ou de la pression de perfusion systolique (PPs) [30, 31]. Une étude montre également une prédominance de la réduction de la PPd la nuit [7]. Dans l’étude Egna-Neumarkt [1], la prévalence du glaucome augmente lorsque la PPd est inférieure à 50 mmHg [risque relatif (RR) de 4,5]. Dans l’étude Proyecto VER [41], le RR est de 3 pour une PPd inférieure à 45 mmHg comparativement à une PPd de 65 mmHg. Dans la Barbados Eye Study [30, 31], la PPd et la prévalence du glaucome sont corrélées, avec un risque relatif de 3,3 chez les sujets ayant une PPd inférieure au 20e percentile. Les sujets ayant une PPd inférieure à 30 mmHg ont un risque de GPAO six fois supérieur à ceux ayant une PPd supérieure à 50 mmHg [53]. Dans la Rotterdam Study [24], le GPAO était associé à une PPd plus basse chez les sujets ayant une HTO et une hypertension artérielle (HTA) traitée. L’association entre une faible PPd et un GPAO a été confirmée chez les patients ayant une HTA [54]. Les RR à neuf ans de GPAO étaient respectivement de 2,1, 2 et 2,6 en cas de faibles valeurs de PPd, PPs ou PPm [31].
Le territoire vasculaire oculaire concerné par ces modifications pathologiques du flux sanguin est de taille variable, localisé à un territoire anatomique précis s’il existe une autorégulation pathologique locale, ou étendu à l’ensemble des structures anatomiques de l’œil dans le cadre d’une diminution importante de la PPm secondaire à des pathologies cardiovasculaires associées. Les caractéristiques du FSO au cours du GPAO sont détaillées en fonction des lits vasculaires (tête du nerf optique, choroïde, rétine).
Les études en échographie Doppler ont montré une diminution de la vélocité au sein de l’AO ainsi qu’une augmentation de l’index de résistance en cas de GPAO, comparativement à des sujets sains [26]. Il existe une corrélation importante entre la vélocité de l’artère centrale de la rétine et la pression artérielle moyenne (PAM) [26]. Les patients glaucomateux présentant une réduction nocturne de la pression artérielle (over-dipper) ont une réduction de la vélocité diastolique de l’AO et une augmentation de l’index de résistance comparativement aux patients sans over-dipping.
Une augmentation des index de résistance (> 0,75) du flux sanguin rétrobulbaire [2] et une diminution de la vélocité diastolique et moyenne [2] sont des facteurs prédictifs de conversion vers un GPAO. Par ailleurs, les patients se dégradant sur le plan pémétrique ont une plus faible vélocité diastolique et un index de résistance de l’AO plus élevé que les patients stables. Chez ces patients, il existe également une corrélation positive entre la PPm et la vélocité diastolique de l’AO et de l’artère centrale de la rétine, et une corrélation négative entre PPm et l’index de résistance de l’AO [19]. La vélocité diastolique de l’artère centrale de la rétine [43] et des artères ciliaires postérieures [3] est également négativement corrélée à l’aggravation du champ visuel. Les paramètres du flux sanguin nocturne de l’AO sont normaux chez les patients présentant une stabilité du champ visuel [23]. Les patients présentant une atteinte campimétrique sévère ont une réduction significative des vélocités des vaisseaux rétrobulbaires.
Les patients présentant une asymétrie d’atteinte du champ visuel ont également une asymétrie des vélocités au niveau de l’AO [37, 48] et de l’artère centrale de la rétine [37]. La régulation vasculaire au sein des vaisseaux rétrobulbaires et/ou de l’artère centrale de la rétine est anormale lors des changements de posture (augmentation de vélocité) et pendant l’hypercapnie. La réponse des vaisseaux rétrobulbaires à l’hyperoxie serait conservée.
Chez les patients glaucomateux, le flux sanguin choroïdien au niveau maculaire mesuré par fluxmétrie laser Doppler (LDF, laser Doppler flowmetry) est diminué de 30 % par rapport à celui des sujets sains contrôles [33]. Il n’existe cependant pas de corrélation entre les paramètres du flux (vélocité, volume) et l’atteinte du champ visuel dans les glaucomes débutants [33]. Les données obtenues à l’aide du pulsatile ocular blood flow (POBF), reflet des variations de flux au niveau de l’ensemble de la choroïde, vont également dans le même sens (– 14 à – 22 %) [27]. Il existe en outre une diminution plus sévère du flux de sanguin choroïdien si un syndrome de Raynaud est associé [12].
La capacité de régulation du flux sanguin choroïdien est altérée chez les patients glaucomateux notamment au cours d’une augmentation de la pression artérielle (PA) à l’exercice, entraînant une variabilité plus importante du flux sanguin choroïdien. Le flux sanguin choroïdien est également diminué lors d’un exercice isométrique chez les patients atteints de GPAO ayant un vasospasme, ce qui suggère une réduction de l’autorégulation dans ce contexte. Le POBF n’est pas corrélé au flux sanguin rétinien parapapillaire chez les patients présentant un GPAO [10].
Le flux sanguin mesuré par LDF au niveau de la tête du nerf optique (TNO) est réduit de 30 % dans le GPAO [36]. La LDF détecte des anomalies précoces du flux sanguin vasculaire au niveau de la TNO chez les patients suspects de GPAO avant l’apparition de déficit du champ visuel [35], ce qui laisse penser que l’altération du flux sanguin serait une des causes de la progression de la maladie. Une altération de l’autorégulation du flux sanguin de la TNO lors de stimulation lumineuse intermittente (flicker) est détectée à la phase précoce du glaucome [46], suggérant une atteinte du couplage neurovasculaire au sein de la rétine. À l’inverse, pour une augmentation modérée de la pression intra-oculaire (PIO), l’autorégulation au niveau de la TNO est maintenue [56].
D’autres arguments liés à la survenue du glaucome ou à sa progression sont en faveur d’une relation entre le flux sanguin de la TNO et le glaucome. Premièrement, la relation topographique : la diminution du flux sanguin de la TNO des sujets ayant une HTO est inversement proportionnelle à leur rapport cup/disc avant l’apparition de déficit du champ visuel ; la réduction du flux sanguin est plus importante au niveau de l’anneau neurorétinien temporal [27, 36]. Deuxièmement, la relation de sévérité : il existe une corrélation entre les régions avec faible flux sanguin de la TNO, les régions d’excavation de la papille, la topographie et la profondeur des déficits périmétriques [36]. La progression des déficits du champ visuel est plus rapide en cas de diminution du volume sanguin au niveau de la TNO [57]. Les lésions glaucomateuses sévères sont associées à une diminution du flux sanguin au niveau de la lame criblée et au niveau de l’anneau neurorétinien [15, 22]. Au cours des variations nycthémérales de la PPm/PIO dans le GPAO non traité, les variations du flux de la TNO sont plus importantes dans les régions atteintes de l’anneau neurorétinien [49].
Une diminution importante du flux sanguin de la TNO est fréquemment associée à une dysrégulation vasculaire digitale au cours d’un phénomène de Raynaud [12]. En dehors d’un contexte de vasospasme, les patients glaucomateux présentent également une diminution significative du flux sanguin de la TNO lors d’un test de provocation à l’eau froide.
Le flux sanguin de la TNO des patients glaucomateux ayant une HTA est plus important comparativement aux patients n’en ayant pas [20], suggérant que le traitement antihypertensif pourrait être délétère pour le flux de la TNO. Une dysrégulation du flux sanguin de la TNO et de la PPm par rapport à la PA est retrouvée chez les sujets GPAO [18].
Il a été mis en évidence par vidéo-angiographie une diminution plus marquée du flux sanguin rétinien par rapport au flux sanguin choroïdien dans le GPAO. Cependant, il n’a pas été retrouvé de corrélation significative entre le flux sanguin parapapillaire et la sévérité de la perte du champ visuel [9]. Les lésions glaucomateuses sévères ne sont pas associées à une diminution du flux sanguin au niveau de la rétine péripapillaire, mais à une plus grande variabilité du flux sanguin parapapillaire [9].
Un rétrécissement des artérioles et veinules rétiniennes est associé au glaucome avec un RR de 1,2. Il existe une diminution du flux sanguin rétinien (rétinographe Heidelberg) [32] équivalente chez les patients avec GPAO et glaucome à pression normale (GPN) [32], et corrélée aux régions les plus atteintes au niveau du nerf optique [32]. Des mesures du flux sanguin rétinien en OCT montrent une réduction du flux sanguin rétinien total et une bonne corrélation entre la réduction du flux sanguin et les atteintes du champ visuel [25]. Les anomalies structurales et celles du flux sanguin rétinien sont indépendantes, suggérant que ces deux paramètres sont complémentaires pour prédire l’atteinte fonctionnelle.
Le glaucome est également associé à une réduction de la vasoréactivité rétinienne en hypercapnie [55] lors du changement de posture [14] et de l’augmentation transitoire de la PIO [21, 34]. Des micro-irrégularités des parois des branches de l’artère centrale de la rétine analysées à l’aide du retinal vessel analyzer (RVA) sont observées en cas de GPAO lors des épisodes de dilatation vasculaire, suggérant une dysfonction endothéliale [29]. La vasoréactivité des vaisseaux rétiniens (moindre vasodilatation des veines) est également retrouvée après stimulation flicker (couplage neurovasculaire). Une vasoconstriction péripapillaire des artères rétiniennes a été décrite chez 42 % des patients avec GPAO [42]. La résistance de l’artère centrale de la rétine est augmentée chez ces patients ayant un over-dip de leur PA nocturne. Avec la technique de LDF, il n’a pas été mis en évidence de différence de flux sanguin fovéolaire entre les patients avec GPAO et les patients contrôles.
Un syndrome vasospastique doit être recherché chez les patients ayant un GPAO. L’association est retrouvée chez environ 25 % d’entre eux. La vasodilatation dépendant de l’endothélium, explorée au niveau de l’avant-bras, est significativement plus faible chez les patients ayant un GPAO comparativement à des sujets normaux [50]. Une dysfonction endothéliale systémique associée à une élévation du taux d’endothéline 1 est plus fréquente, et le taux d’endothéline plasmatique est corrélé à la progression des déficits du champ visuel chez les patients avec GPAO (contrairement aux patients avec GPN) [4, 13]. La concentration d’endothéline dans l’humeur aqueuse est augmentée d’environ 30 %. Les études publiées concernant les dosages d’endothéline doivent être considérées avec prudence, car les techniques de dosage utilisées ne sont pas validées et leurs résultats sont parfois contradictoires selon les caractéristiques démographiques des populations étudiées. Le rôle des récepteurs de l’endothéline reste encore à préciser, l’effet d’un inhibiteur des récepteurs de l’endothéline sur le flux sanguin rétinien, choroïdien et de la TNO étant similaire chez le patient glaucomateux et le sujet normal.
Il a été également décrit une réduction du flux sanguin cérébral et une absence de vasoréactivité cérébrale lors de l’inhalation d’oxygène (habituellement vasoconstrictrice).
Une réduction de la PPm nocturne plus basse est rapportée chez les patients ayant un GPN comparativement à un groupe contrôle, et est corrélée à un allongement du temps de transit artérioveineux [38]. L’amplitude de la PPm mesurée sur 24 heures n’est pas corrélée aux amplitudes de la PIO chez les patients avec GPN [45]. Ces patients over-dippers nocturnes ont également des fluctuations de la PPm plus importantes que les dippers physiologiques. Une réduction de la PPm est associée à une progression des déficits du champ visuel chez les patients avec GPN traités [8]. Cependant, des données contradictoires existent concernant la relation entre les fluctuations de la PPm et l’aggravation du champ visuel [45].
Les études retrouvent une altération du FSO chez les patients ayant un GPN. Cette altération de la fonction vasculaire oculaire a été identifiée dans différents territoires anatomiques oculaires [52].
L’ étude en échographie Doppler montre une diminution de la vélocité du flux sanguin ainsi qu’une augmentation de l’index de résistance de l’AO chez les patients avec GPN comparativement aux sujets sains [44]. Ces modifications sont également retrouvées au sein des artères ciliaires courtes postérieures [44] ou de l’artère centrale de la rétine. Les paramètres hémodynamiques sont altérés dans les yeux les plus sévèrement atteints (champ visuel) [28].
La réponse du flux ophtalmique lors d’une augmentation de la pCO2 est anormale (augmentation de la vélocité), suggérant l’effet vasodilatateur du CO2 sur l’AO présentant un tonus vasoconstricteur. L’atteinte de l’autorégulation lors de changement de posture aboutit à une augmentation de la vélocité dans les artères ciliaires postérieures.
Évalué par POBF ou angiographie, le flux sanguin choroïdien est réduit dans le GPN par rapport à des sujets sains [17, 40]. Les patients ayant un champ visuel asymétrique ont un flux sanguin choroïdien plus réduit du côté présentant les déficits les plus importants [17].
Le flux sanguin de la TNO est significativement diminué par rapport aux sujets sains [32] et serait associé à la PA. L’altération du flux sanguin est associée aux dommages structurels de l’anneau neurorétinien [32] et aux déficits fonctionnels du champ visuel [47]. L’autorégulation du flux sanguin de la TNO lors de modifications de la PPm ne semble pas altérée chez ces patients, bien qu’il existe une augmentation locale des résistances vasculaires [39].
Une évaluation du temps de transit du flux sanguin rétinien par vidéo-angiographie à la fluorescéine ne montre pas de différence entre les groupes GPAO et GPN. Cependant, le flux sanguin rétinien parapapillaire mesuré par LDF chez les patients ayant un GPN est réduit par rapport à un groupe contrôle de sujets sains [6]. Un plus petit diamètre des vaisseaux rétiniens est associé à une réduction plus importante de la couche des fibres nerveuses rétiniennes [5]. L’analyse de la vitesse au sein des vaisseaux rétiniens par vélocimétrie laser Doppler objective une réduction de la vitesse associée à des anomalies rhéologiques.
Un syndrome vasospastique doit être recherché chez les patients ayant un GPN. Une association avec le GPN est retrouvée chez environ 50 % des patients [16].
La fonction endothéliale périphérique est altérée (réponse du flux sanguin de l’avant-bras à l’injection d’acétylcholine, vasodilatatrice), avec une balance vasodilatation/vasoconstriction déséquilibrée en faveur de la vasoconstriction périphérique. La vasodilatation dépendant de l’endothélium, explorée au niveau de l’avant-bras, est significativement plus faible chez les patients avec GPN que chez les patients avec GPAO ou normaux [51]. La dysfonction endothéliale à l’endothéline 1 (vasoconstriction exagérée) et à la 5-hydroxy-tryptamine (vasodilatation exagérée) a été confirmée sur des prélèvements vasculaires issus de graisse sous-cutanée. Il n’est cependant pas démontré que ces dysrégulations existent au sein du lit vasculaire oculaire ou de l’AO.
Les patients ayant un GPN ont une réponse excessive de la vasoconstriction distale au froid [11], la durée de cette vasoconstriction étant plus longue.
L’augmentation du taux d’endothéline plasmatique comparativement aux sujets sains n’est pas formellement démontrée, les études étant contradictoires. Il n’existe pas de corrélation entre le taux d’endothéline plasmatique et les déficits du champ visuel. Le système endothéline mérite d’être exploré à l’avenir, car les dosages plasmatiques d’endothéline des études antérieures présentaient de nombreuses insuffisances méthodologiques.
L’étude des différents lits vasculaires oculaires, assurant la vascularisation de la rétine, de la choroïde et du nerf optique, montre que la survenue d’une neuropathie glaucomateuse est associée, à divers degrés, à une perfusion oculaire anormale (réduction, variabilité) et une dysrégulation vasculaire (moindre autorégulation, vasospasticité, dysfonction endothéliale). Une meilleure caractérisation des réponses vasculaires au décours de la neuropathie optique glaucomateuse permettra d’identifier les cibles thérapeutiques permettant d’améliorer la perfusion oculaire et/ou de corriger les dysrégulations vasculaires qui y sont associées.
Retenir
La physiopathologie exacte du glaucome n’est pas parfaitement élucidée.
Une diminution de la PPm et une perte de l’autorégulation vasculaire sont incriminées dans la pathologie glaucomateuse.
Une diminution du FSO a été identifiée dans le GPAO et le GPN. Cette diminution est précoce et contribue à la progression de la neuropathie optique.
Le clinicien devra toujours s’efforcer d’identifier et de traiter ces facteurs de risque vasculaires potentiellement responsables d’une aggravation du GPAO.
[1] Bonomi L, Marchini G, Marraffa M, et al. Vascular risk factors for primary open angle glaucoma : the Egna-Neumarkt Study. Ophthalmology. 2000 ; 107 : 1287-93.
[2] Calvo P, Ferreras A, Polo V, et al. Predictive value of retrobulbar blood flow velocities in glaucoma suspects. Invest Ophthalmol Vis Sci. 2012 ; 53 : 3875-84.
[3] Cellini M, Possati GL, Sbrocca M, Caramazza N. Correlation between visual field and color Doppler parameters in chronic open angle glaucoma. Int Ophthalmol. 1996 ; 20 : 215-9.
[4] Cellini M, Strobbe E, Gizzi C, et al. Endothelin-1 plasma levels and vascular endothelial dysfunction in primary open angle glaucoma. Life Sci. 2012 ; 91 : 699-702.
[5] Chang M, Yoo C, Kim SW, Kim YY. Retinal vessel diameter, retinal nerve fiber layer thickness, and intraocular pressure in korean patients with normal-tension glaucoma. Am J Ophthalmol. 2011 ; 151 : 100-5.
[6] Chung HS, Harris A, Kagemann L, Martin B. Peripapillary retinal blood flow in normal tension glaucoma. Br J Ophthalmol. 1999 ; 83 : 466-9.
[7] Costa VP, Jimenez-Roman J, Carrasco FG, et al. Twenty-four-hour ocular perfusion pressure in primary open-angle glaucoma. Br J Ophthalmol. 2010 ; 94 : 1291-4.
[8] De Moraes CG, Liebmann JM, Greenfield DS, et al. Risk factors for visual field progression in the low-pressure glaucoma treatment study. Am J Ophthalmol. 2012 ; 154 : 702-11.
[9] Deokule S, Vizzeri G, Boehm A, et al. Association of visual field severity and parapapillary retinal blood flow in open-angle glaucoma. J Glaucoma. 2010 ; 19 : 293-8.
[10] Deokule S, Vizzeri G, Boehm AG, et al. Correlation among choroidal, parapapillary, and retrobulbar vascular parameters in glaucoma. Am J Ophthalmol. 2009 ; 147 : 736-43.
[11] Drance SM, Douglas GR, Wijsman K, et al. Response of blood flow to warm and cold in normal and low-tension glaucoma patients. Am J Ophthalmol. 1988 ; 105 : 35-9.
[12] Emre M, Orgul S, Gugleta K, Flammer J. Ocular blood flow alteration in glaucoma is related to systemic vascular dysregulation. Br J Ophthalmol. 2004 ; 88 : 662-6.
[13] Emre M, Orgul S, Haufschild T, et al. Increased plasma endothelin-1 levels in patients with progressive open angle glaucoma. Br J Ophthalmol. 2005 ; 89 : 60-3.
[14] Feke GT, Pasquale LR. Retinal blood flow response to posture change in glaucoma patients compared with healthy subjects. Ophthalmology. 2008 ; 115 : 246-52.
[15] Findl O, Rainer G, Dallinger S, et al. Assessment of optic disk blood flow in patients with open-angle glaucoma. Am J Ophthalmol. 2000 ; 130 : 589-96.
[16] Flammer J, Mozaffarieh M. What is the present pathogenetic concept of glaucomatous optic neuropathy ? Surv Ophthalmol. 2007 ; 52 (Suppl. 2) : S162-73.
[17] Fontana L, Poinoosawmy D, Bunce CV, et al. Pulsatile ocular blood flow investigation in asymmetric normal tension glaucoma and normal subjects. Br J Ophthalmol. 1998 ; 82 : 731-6.
[18] Fuchsjager-Mayrl G, Wally B, Georgopoulos M, et al. Ocular blood flow and systemic blood pressure in patients with primary open-angle glaucoma and ocular hypertension. Invest Ophthalmol Vis Sci. 2004 ; 45 : 834-9.
[19] Gherghel D, Orgul S, Gugleta K, et al. Relationship between ocular perfusion pressure and retrobulbar blood flow in patients with glaucoma with progressive damage. Am J Ophthalmol. 2000 ; 130 : 597-605.
[20] Grunwald JE, Piltz J, Hariprasad SM, et al. Optic nerve blood flow in glaucoma : effect of systemic hypertension. Am J Ophthalmol. 1999 ; 127 : 516-22.
[21] Grunwald JE, Riva CE, Stone RA, et al. Retinal autoregulation in open-angle glaucoma. Ophthalmology. 1984 ; 91 : 1690-4.
[22] Harju M, Vesti E. Blood flow of the optic nerve head and peripapillary retina in exfoliation syndrome with unilateral glaucoma or ocular hypertension. Graefes Arch Clin Exp Ophthalmol. 2001 ; 239 : 271-7.
[23] Harris A, Spaeth G, Wilson R, et al. Nocturnal ophthalmic arterial hemodynamics in primary open-angle glaucoma. J Glaucoma. 1997 ; 6 : 170-4.
[24] Hulsman CA, Vingerling JR, Hofman A, et al. Blood pressure, arterial stiffness, and open-angle glaucoma : the Rotterdam study. Arch Ophthalmol. 2007 ; 125 : 805-12.
[25] Hwang JC, Konduru R, Zhang X, et al. Relationship among visual field, blood flow, and neural structure measurements in glaucoma. Invest Ophthalmol Vis Sci. 2012 ; 53 : 3020-6.
[26] Kaiser HJ, Schoetzau A, Stumpfig D, Flammer J. Blood-flow velocities of the extraocular vessels in patients with high-tension and normal-tension primary open-angle glaucoma. Am J Ophthalmol. 1997 ; 123 : 320-7.
[27] Kerr J, Nelson P, O’Brien C. A comparison of ocular blood flow in untreated primary open-angle glaucoma and ocular hypertension. Am J Ophthalmol. 1998 ; 126 : 42-51.
[28] Kondo Y, Niwa Y, Yamamoto T, et al. Retrobulbar hemodynamics in normal-tension glaucoma with asymmetric visual field change and asymmetric ocular perfusion pressure. Am J Ophthalmol. 2000 ; 130 : 454-60.
[29] Kotliar KE, Nagel E, Vilser W, Lanzl IM. Functional in vivo assessment of retinal artery microirregularities in glaucoma. Acta Ophthalmol. 2008 ; 86 : 424-33.
[30] Leske MC, Connell AM, Wu SY, et al. Risk factors for open-angle glaucoma. The Barbados Eye Study. Arch Ophthalmol. 1995 ; 113 : 918-24.
[31] Leske MC, Wu SY, Hennis A, et al. Risk factors for incident open-angle glaucoma : the Barbados Eye Studies. Ophthalmology. 2008 ; 115 : 85-93.
[32] Logan JF, Rankin SJ, Jackson AJ. Retinal blood flow measurements and neuroretinal rim damage in glaucoma. Br J Ophthalmol. 2004 ; 88 : 1049-54.
[33] Marangoni D, Falsini B, Colotto A, et al. Subfoveal choroidal blood flow and central retinal function in early glaucoma. Acta Ophthalmol. 2012 ; 90 : e288-94.
[34] Nagel E, Vilser W, Lanzl IM. Retinal vessel reaction to short-term IOP elevation in ocular hypertensive and glaucoma patients. Eur J Ophthalmol. 2001 ; 11 : 338-44.
[35] Piltz-seymour JR, Grunwald JE, Hariprasad SM, Dupont J. Optic nerve blood flow is diminished in eyes of primary open-angle glaucoma suspects. Am J Ophthalmol. 2001 ; 132 : 63-9.
[36] Piltz-Seymour JR. Laser Doppler flowmetry of the optic nerve head in glaucoma. Surv Ophthalmol. 1999 ; 43 (Suppl. 1) : S191-8.
[37] Plange N, Kaup M, Arend O, Remky A. Asymmetric visual field loss and retrobulbar haemodynamics in primary open-angle glaucoma. Graefes Arch Clin Exp Ophthalmol. 2006 ; 244 : 978-83.
[38] Plange N, Kaup M, Remky A, Arend KO. Prolonged retinal arteriovenous passage time is correlated to ocular perfusion pressure in normal tension glaucoma. Graefes Arch Clin Exp Ophthalmol. 2008 ; 246 : 1147-52.
[39] Pournaras CJ, Riva CE, Bresson-Dumont H, et al. Regulation of optic nerve head blood flow in normal tension glaucoma patients. Eur J Ophthalmol. 2004 ; 14 : 226-35.
[40] Prünte C. Indocyanine green angiography in patients with normal-tension glaucoma. In : Kaiser HJ, Hendrickson P (eds). Ocular blood flow, new insights into the pathogenesis of ocular diseases. Basel, Karger, 1996 : 189-94.
[41] Quigley HA, West SK, Rodriguez J, et al. The prevalence of glaucoma in a population-based study of Hispanic subjects : Proyecto VER. Arch Ophthalmol. 2001 ; 119 : 1819-26.
[42] Rader J, Feuer WJ, Anderson DR. Peripapillary vasoconstriction in the glaucomas and the anterior ischemic optic neuropathies. Am J Ophthalmol. 1994 ; 117 : 72-80.
[43] Rankin SJ, Drance SM. Peripapillary focal retinal arteriolar narrowing in open angle glaucoma. J Glaucoma. 1996 ; 5 : 22-8.
[44] Rankin SJ. Color Doppler imaging of the retrobulbar circulation in glaucoma. Surv Ophthalmol. 1999 ; 43 (Suppl. 1) : S176-82.
[45] Renard E, Palombi K, Gronfier C, et al. Twenty-four hour (Nyctohemeral) rhythm of intraocular pressure and ocular perfusion pressure in normal-tension glaucoma. Invest Ophthalmol Vis Sci. 2009 ; 51 : 882-9.
[46] Riva CE, Salgarello T, Logean E, et al. Flicker-evoked response measured at the optic disc rim is reduced in ocular hypertension and early glaucoma. Invest Ophthalmol Vis Sci. 2004 ; 45 : 3662-8.
[47] Sato EA, Ohtake Y, Shinoda K, et al. Decreased blood flow at neuroretinal rim of optic nerve head corresponds with visual field deficit in eyes with normal tension glaucoma. Graefes Arch Clin Exp Ophthalmol. 2006 ; 244 : 795-801.
[48] Schumann J, Orgul S, Gugleta K, et al. Interocular difference in progression of glaucoma correlates with interocular differences in retrobulbar circulation. Am J Ophthalmol. 2000 ; 129 : 728-33.
[49] Sehi M, Flanagan JG, Zeng L, et al. Anterior optic nerve capillary blood flow response to diurnal variation of mean ocular perfusion pressure in early untreated primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2005 ; 46 : 4581-7.
[50] Su WW, Cheng ST, Ho WJ, et al. Glaucoma is associated with peripheral vascular endothelial dysfunction. Ophthalmology. 2008 ; 115 : 1173-8.
[51] Su WW, Cheng ST, Hsu TS, Ho WJ. Abnormal flow-mediated vasodilation in normal-tension glaucoma using a noninvasive determination for peripheral endothelial dysfunction. Invest Ophthalmol Vis Sci. 2006 ; 47 : 3390-4.
[52] Sugiyama T, Schwartz B, Takamoto T, Azuma I. Evaluation of the circulation in the retina, peripapillary choroid and optic disk in normal-tension glaucoma. Ophthalmic Res. 2000 ; 32 : 79-86.
[53] Tielsch JM, Katz J, Sommer A, et al. Hypertension, perfusion pressure, and primary open-angle glaucoma. A population-based assessment. Arch Ophthalmol. 1995 ; 113 : 216-21.
[54] Topouzis F, Wilson MR, Harris A, et al. Association of open-angle glaucoma with perfusion pressure status in the Thessaloniki Eye Study. Am J Ophthalmol. 2013 ; 155 : 843-51.
[55] Venkataraman ST, Hudson C, Rachmiel R, et al. Retinal arteriolar vascular reactivity in untreated and progressive primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2010 ; 51 : 2043-50.
[56] Weigert G, Findl O, Luksch A, et al. Effects of moderate changes in intraocular pressure on ocular hemodynamics in patients with primary open-angle glaucoma and healthy controls. Ophthalmology. 2005 ; 112 : 1337-42.
[57] Zink JM, Grunwald JE, Piltz-Seymour J, et al. Association between lower optic nerve laser Doppler blood volume measurements and glaucomatous visual field progression. Br J Ophthalmol. 2003 ; 87 : 1487-91.
J.-P. Renard, H. El Chehab, M. Francoz
Nos connaissances ont nettement progressé dans le domaine des facteurs physiopathogéniques de la neuropathie optique glaucomateuse. Elle est caractérisée par un certain nombre de facteurs de risque (FDR), mécaniques et vasculaires mais aussi génétiques, épigénétiques et environnementaux, dont la fréquence et le degré d’expression variables expliquent la variété des formes cliniques de l’affection.
Les résultats des grandes études épidémiologiques et cliniques ont permis une meilleure connaissance des caractéristiques de certains FDR cliniques de développement, d’apparition et de progression du glaucome primitif à angle ouvert (GPAO). Leur identification reste importante même si le consensus sur leur identité n’est pas absolu.
Le GPAO est caractérisé également par un certain nombre de particularités génétiques hétérogènes. La prévalence de l’affection est nettement plus élevée en cas de GPAO chez des parents du premier degré. Une mutation de certains gènes, impliqués notamment dans le métabolisme du collagène du stroma cornéen et de la membrane de Descemet, pourrait intervenir sur l’épaisseur cornéenne, alors que d’autres mutations influeraient sur le déterminisme de différents paramètres cliniques (taille du disque optique, etc.) [1, 15, 22, 23].
Les particularités des facteurs génétiques épigénétiques et environnementaux doivent être précisées.
J.-P. Renard, M. Francoz
Le GPAO chez les enfants et les adultes jeunes répond à une génétique mendélienne à la différence du GPAO de l’adulte âgé.
Dans le GPAO de l’adulte, de nombreuses études d’association du génome ont permis d’isoler certains des gènes de susceptibilité impliqués dans les variations des paramètres cliniques habituellement pris en considération dans l’évaluation des patients glaucomateux (disque optique, épaisseur cornéenne centrale, etc.).
Les facteurs épigénétiques impliqués dans les maladies neurodégénératives semblent également jouer un rôle important dans le GPAO.
L’importance des antécédents familiaux ainsi que des facteurs ethniques est mieux précisée grâce aux données des récentes méta-analyses de l’ensemble des études réalisées à ce jour.
Le caractère hétérogène des particularités génétiques et cliniques du GPAO est bien établi.
Les gènes impliqués dans la physiopathologie du glaucome sont désignés dans la nomenclature internationale par « Human Genome Organization/Genome Database » et le symbole « GLC ». Le chiffre qui suit (1, 2 ou 3) représente respectivement le GPAO, le glaucome à angle fermé et les sous-types congénital et juvénile du glaucome à angle ouvert. Enfin les lettres A, B, C, etc., désignent leur ordre de découverte. Par exemple, GLC1A correspond au gène MYOC dont la protéine est la myociline [10].
Le GPAO chez les enfants et les adultes jeunes répond à une génétique mendélienne à la différence du GPAO de l’adulte âgé. En effet, seuls quatre gènes ont été incriminés pour une transmission mendélienne, le gène GLC1A de la myociline, le gène GLC1E de l’optineurine, le gène GLC1G de la protéine WDR36 et le gène GLC1O de la neutrophine 4 (NTF4) (voir chapitre 4). Ces quatre gènes seraient à l’origine de moins de 5 % des GPAO. Leur mutation entraîne soit une altération au niveau trabéculaire (mutation du gène de la myociline), soit une susceptibilité plus importante des cellules ganglionnaires à l’apoptose cellulaire (mutation du gène de NTF4). La transmission est autosomique dominante à pénétrance variable [5, 10].
La cause la plus fréquemment retrouvée de GPAO juvénile parmi la plupart des populations reste la mutation MYOC hétérozygote (autosomique dominante) que l’on retrouve jusque dans un tiers des cas et qui peut être impliquée parfois dans la survenue de glaucome plus précoce et de glaucome tardif.
Bien que le GPAO de l’adulte, d’étiologie complexe, ne suive en général pas une simple génétique mendélienne, plusieurs études rapportent des facteurs de susceptibilité génétique. Dans quelques cas, la survenue du GPAO de l’adulte peut être liée à une mutation hétérozygote des gènes MYOC, OPTN ou WDR6. Plus récemment, une mutation hétérozygote du gène NTF4 a été retrouvée associée au phénotype d’un petit pourcentage de patients au sein d’une cohorte de sujets allemands [10].
La majeure partie des autres cas de GPAO de l’adulte serait liée à une action combinée d’une susceptibilité génétique influencée par une action de l’environnement (voir plus loin les facteurs épigénétiques). Depuis près de deux décennies, de nombreuses études d’association du génome ont permis d’isoler certains de ces gènes de susceptibilité. En général, elles comparent des marqueurs de l’ADN, marqueurs du polymorphisme génique, entre une population témoin et une population cible. Ces marqueurs du polymorphisme génique ont une implication dans les variations des paramètres cliniques habituellement pris en considération dans l’évaluation des patients glaucomateux : épaisseur cornéenne centrale, paramètres de la tête du nerf optique (aire du disque optique et excavation verticale), etc. [3, 17]. En ce qui concerne les paramètres papillaires, plusieurs études ont rapporté que des polymorphismes de certains gènes (SIX1, CDKN2BAS, etc.) sont associés de façon significative à une excavation verticale plus importante [13]. Ces variants géniques impliqués, qui peuvent intervenir lors du développement oculaire ou lors de sa croissance, ont été retrouvés comme FDR augmenté de GPAO [16]. D’autres études ont également identifié le locus CDKN2BAS comme facteur de susceptibilité génétique dans d’autres affections liées à l’âge (insuffisance coronarienne, anévrisme intracrânien et diabète de type 2). Il intervient également dans le codage d’un ARN régulateur de l’expression d’un composant (CDKN2B) du facteur de croissance TGF-β (transforming growth factor-β) dont le rôle a été rapporté au niveau du nerf optique et de la voie d’écoulement trabéculaire dans le glaucome [25]. Ces données laissent entrevoir des perspectives intéressantes de recherche dans le développement de stratégies de neuroprotection dans le glaucome.
Les facteurs épigénétiques régulent l’expression et la fonction des gènes sans modifier la séquence de l’ADN primaire. Les modifications épigénétiques reconnues concernent notamment les composants des noyaux protéiques des nucléosomes (contenant l’ADN) dans les cellules eucaryotes. Les phénomènes épigénétiques se sont révélés des médiateurs importants dans le développement normal et le vieillissement, avec un rôle probable dans les mécanismes physiopathologiques et la progression des maladies liées à l’âge. Plusieurs études rapportent leur rôle dans le développement des cancers et de certaines maladies neurodégénératives chroniques comme la maladie d’Alzheimer [9].
Un certain nombre de facteurs environnementaux comme le régime alimentaire, le tabagisme et la pollution peuvent influer sur la régulation épigénétique de l’expression des gènes. Il semble que la région génomique du locus CDKN2BAS récemment associée au GPAO et au rapport c/d du nerf optique soit régulée par les mécanismes épigénétiques [13, 26].
Les études concernant l’implication des effets épigénétiques dans le glaucome en sont à leurs premiers stades. Elles pourraient avoir un impact significatif sur les approches thérapeutiques à venir.
Au total, les FDR génétiques du GPAO restent complexes. Ils expliquent la grande diversité des présentations cliniques de la neuropathie. La relation entre la pression intra-oculaire (PIO) et l’atteinte des cellules ganglionnaires n’est pas univoque. En effet, certains patients peuvent présenter une hypertonie oculaire (HTO) sans atteinte structurelle ou fonctionnelle, alors que d’autres développent des atteintes glaucomateuses avec une PIO normale [25].
Dans nos pays occidentaux, la mutation autosomique dominante MYOC représente une cause courante de GPAO juvénile. Elle est peu fréquemment impliquée dans la survenue des formes cliniques chez l’adulte. L’identification de telles familles pourrait bénéficier de conseil génétique. La survenue du GPAO de l’adulte suit rarement une simple transmission génétique mendélienne, mais les études du polymorphisme génique (études génomiques) réalisées au sein de différentes populations ont mis en évidence un certain nombre de FDR génétiques potentiels pour le phénotype. L’identification des modifications des séquences d’ADN (single nucleotide polymorphisms – SNP), associées au GPAO, permettrait la mise au point de tests diagnostiques de dépistage des sujets à risque (fig. 8-11) [7].
Si nos connaissances progressent avec des perspectives enthousiasmantes, la détermination de phénotypes bien caractérisés doit encore être précisée (voir chapitre 4).

Fig. 8-11 Pathogénie du GPAO : gènes, facteurs environnementaux et effets épigénétiques. (D’après Wiggs et al., 2012 [25].)
La notion de glaucome héréditaire est connue depuis 1869 grâce à Von Graefe [6]. En 1941, Duke-Elder [4] a décrit un type de glaucome appelé glaucome familial, remarqué par sa transmission de type autosomique dominante.
De nombreuses études épidémiologiques et cliniques ont rapporté l’importance des antécédents familiaux et leur rôle prédisposant fortement l’apparition d’un glaucome primitif à angle ouvert [11, 18].
Dans l’étude Baltimore Eye Survey portant sur 161 cas de GPAO, le risque pour les membres de la famille de développer un GPAO a été retrouvé chez les frères et sœurs (OR : 3,69) et les enfants (OR : 1,12) des patients. Il est intéressant de noter que les OR étaient retrouvés supérieurs chez les patients connaissant leur diagnostic de glaucome par rapport à ceux qui l’apprenaient pour la première fois. Il s’agit d’un biais à connaître pour les études sur le FDR constitué par les antécédents familiaux [19]. Les autres grandes études épidémiologiques rapportent des données superposables.
Une méta-analyse récente de l’ensemble des études concernant les facteurs de risque du GPAO, réalisées de janvier 1950 à janvier 2013, permet d’obtenir une synthèse intéressante de l’ensemble des travaux réalisés à ce jour.
Elle regroupe les résultats de 41 études séparées, dont 34 études randomisées, avec un niveau de preuves élevé (qualité de niveaux I et II). Ces dernières ont été incluses dans une analyse primaire.
Cette méta-analyse rapporte une prévalence estimée de GPAO fondée sur ces études de niveaux I et II (soit plus de 86 000 participants) de 2,6 % avec un intervalle de confiance très étroit (IC95 % : 2,1-3,1) malgré une hétérogénéité statistique et bien qu’il n’y ait pas de différence dans la prévalence entre les études de niveau I et celles de niveau II (2,5 % versus 3,2 %). La prévalence du GPAO parmi les patients ayant une PIO élevée (supérieure à 21 mmHg) et une PIO normale (inférieure ou égale à 21 mmHg) a été rapportée dans 18 études disponibles (n = 53 349) et était respectivement de 2,4 % (IC95 % : 1,9-2,9) et 1,6 % (IC95% : 1,2-2,1).
L’augmentation de l’âge est associée à un risque significativement plus haut de GPAO, les patients âgés de plus 80 ans ayant le plus haut risque (prévalence : 7,8 % ; OR : 2,9 ; IC95 % : 1,9-4,3).
Les patients avec un antécédent familial de GPAO (patient du premier degré) ont une fréquence augmentée de GPAO (avec un taux de prévalence de 7,6 %) et un risque relatif OR : 3,3 (IC95 % : 2-5,6) pour l’ensemble des neuf études ayant analysé ce facteur [8].
Dans une population comparable à une population européenne, une étude de la large base de données de l’Utah (3 391 GPAO) a retrouvé aussi des risques relatifs (RR) augmentés de GPAO chez des apparentés du deuxième degré. Le RR était retrouvé chez les cousins du premier degré à 1,45 (IC95 % : 1,16-1,8), mais aussi chez les cousins du second degré à 1,19 (IC95 % : 1,08-1,32) [24]. Dans cette étude, le risque général attribuable aux facteurs familiaux a été retrouvé de 0,20, indiquant que 20 % du risque de GPAO serait d’origine génétique.
En plus du GPAO lui-même, les FDR tels que l’HTO et l’excavation papillaire semblent avoir un caractère familial. Les données de la récente méta-analyse disponible rapportent à ce sujet un certain nombre d’éléments intéressants :
un rapport de l’excavation (C/D) supérieur ou égal à 0,7 était le seuil évalué dans la plupart des études. Les patients dont le C/D était supérieur avaient un risque probable positif résumé de 14 (IC95% : 5,3-39). Les patients avec un rapport C/D inférieur à 0,4 étaient moins fréquemment atteints de glaucome ;
une asymétrie du rapport C/D d’au moins 0,3 était le facteur le plus fréquemment étudié avec une probabilité de glaucome nettement augmentée [8].
En pratique, nous pouvons donc retenir que l’existence d’antécédent familial de GPAO est un facteur fortement prédisposant à la survenue de glaucome. Il représente globalement un facteur de risque de GPAO multiplié par trois en cas de glaucome chez un parent du premier degré. Ainsi, un antécédent familial de GPAO est un facteur de risque indéniable en cas de parenté au premier degré, et en fait un élément indispensable à rechercher à l’interrogatoire.
Les patients mélanodermes présentent une incidence plus importante de GPAO et un taux de cécité supérieure par rapport aux patients caucasiens. L’augmentation de la prévalence du GPAO dans cette population versus une population caucasienne est estimée entre trois à six fois [14].
Cette population présente plusieurs facteurs de prédisposition :
une PIO moyenne plus élevée (18,7 ± 5,2 mmHg versus 16,5 ± 3,0 mmHg) [12] ;
une épaisseur cornéenne centrale plus fine, sous-estimant cette PIO (524,8 ± 38,4 μm versus 544,7 ± 31 μm) [2] ;
des paramètres de la tête du nerf optique particuliers augmentant le risque d’altération (taille du disque optique importante, faible épaisseur des fibres nerveuses péripapillaires et profondeur de l’excavation plus importante) [2, 20]. Ces différents paramètres papillaires engendreraient des forces mécaniques augmentées et localisées sur la tête du nerf optique [14].
Des facteurs économiques, géographiques et culturels peuvent s’associer à ces facteurs de prédisposition anatomiques rendant le diagnostic souvent plus tardif. Ce diagnostic tardif retarde la prise en charge de ces patients pour lesquels l’apparition de la pathologie est plus précoce et l’évolution vers la cécité plus rapide [14].
La Los Angeles Latino Eye Study rapporte ainsi un risque intermédiaire dans cette population, chez laquelle le risque serait multiplié par deux par rapport à la population caucasienne [21].
Cependant, la récente méta-analyse concernant les FDR avec les études à niveau de preuve élevé retrouve chez les patients de race noire une prévalence de l’ordre de 7,5 %, avec un RR de 2,9 (IC95 % : 1,4-5,9) [2].
En pratique, nous pouvons donc retenir un risque de GPAO globalement multiplié par trois chez les patients mélanodermes. Ce FDR d’apparition n’est pas rapporté comme facteur de progression du GPAO dans les résultats des études à niveau de preuve élevé.
Les facteurs héréditaires jouent donc un rôle important dans la physiopathogénie du GPAO. Si les facteurs génétiques interviennent essentiellement dans les GPAO juvéniles, la découverte de plusieurs facteurs de susceptibilité génétique dans le déterminisme de paramètres cliniques suivis au cours de la neuropathie optique glaucomateuse souligne leur importance dans le GPAO de l’adulte. Celle-ci est renforcée par l’effet des facteurs épigénétiques dont nous percevons désormais l’importance dans la physiopathologie des maladies liées au vieillissement.
Tous ces éléments soulignent l’implication de facteurs héréditaires sur les nombreux FDR du GPAO (âge, facteurs oculaires, facteurs environnementaux, etc.).
Retenir
Les facteurs génétiques sont surtout impliqués dans le glaucome juvénile, rarement dans le GPAO de l’adulte.
Plusieurs facteurs de susceptibilité génétique impliqués dans les paramètres cliniques du GPAO (épaisseur cornéenne centrale, rapport C/D, etc.) sont mieux connus.
Rôle régulateur de l’expression et de la fonction des gènes par les facteurs épigénétiques.
Les facteurs impliqués dans le développement du GPAO et la mort des cellules ganglionnaires rétiniennes sont multiples.
Ces facteurs incluent des modifications des séquences d’ADN (SNP) et des facteurs environnementaux.
[1] Desronvil T, Logan-Wyatt D, Abdrabou W, et al Distribution of COL8A2 and COL8A1 genes variants in Caucasian primary open angle glaucoma patients with thin central corneal thickness. Mol Vis. 2010 ; 16 : 2185-91.
[2] Dimasi DP, Burdon KP, Craig JE. The genetics of central corneal thickness. Br J Ophthalmol. 2010 ; 94 : 971-6.
[3] Dimasi DP, Burdon KP, Hewitt AW, et al. Genetic investigation into the endophenotypic status of central corneal thickness and optic disc parameters in relation to open-angle glaucoma. Am J Ophthalmol. 2012 ; 154 : 833-42.
[4] Duke-Elder. Textbook of ophthalmology. St Louis, Mosby, 1941.
[5] Fingert JH. Primary open-angle glaucoma genes. Eye Lond Engl. 2011 ; 25 : 587-95.
[6] Graefe V. Beitrage zur pathologie und therapie des glaukoms. Arch Ophthalmol. 1869 ; 15 : 108-252
[7] He S, Li X, Chan N, et al. Review : Epigenetics mechanisms in ocular disease. Mol Vis. 2013 ; 19 : 665-74.
[8] Hollands H, Johnson D, Hollands S, et al. Do findings on routine examination identify patients at risk for primary open-angle glaucoma ? JAMA. 2013 ; 309 : 2035-42.
[9] Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012 ; 81 : 303-11. Qureshi IA, Mehler MF. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep. 2011 ; 11 : 464-73.
[10] Khan AO. Genetics of primary glaucoma. Curr Opin Ophthalmol. 2011 ; 22 : 347-55.
[11] Le A, Mukesh BN, McCarty CA, Taylor HR. Risk factors associated with the incidence of open-angle glaucoma : the visual impairment project. Invest Ophthalmol Vis Sci. 2003 ; 44 : 3783-9.
[12] Leske MC, Connell AM, Wu SY, et al. Distribution of intraocular pressure. The Barbados Eye Study. Arch Ophthalmol. 1997 ; 115 : 1051-7.
[13] Mabuchi F, Sakurada Y, Kashiwagi K, et al. Association between genetic variants associated with vertical cup-to-disc ratio and phenotypic features of primary open-angle glaucoma. Ophthalmology. 2012 ; 119 : 1819-25.
[14] Racette L, Wilson MR, Zangwill LM, et al. Primary open-angle glaucoma in blacks : a review. Surv Ophthalmol. 2003 ; 48 : 295-313.
[15] Ramdas WD, van Koolwijk LM, Ikram MK, et al. A genome-wide association study of optic disc parameters. Plos Genet. 2010 ; 6 : e1000978.
[16] Ramdas WD, van Koolwijk LME, Lemij HG, et al. Common genetic variants associated with open-angle glaucoma. Hum Mol Genet. 2011 ; 20 : 2464-71.
[17] Scheetz TE, Fingert JH, Wang K, et al. A genome-wide association study for primary open angle glaucoma and macular degeneration reveals novel Loci. Plos One. 2013 ; 8 : e58657.
[18] Sommer A. Glaucoma risk factors observed in the Baltimore Eye Survey. Curr Opin Ophthalmol. 1996 ; 7 : 93-8.
[19] Tielsch JM, Katz J, Sommer A, et al. Family history and risk of primary open angle glaucoma. The Baltimore Eye Survey. Arch Ophthalmol. 1994 ; 112 : 69-73.
[20] Tjon-Fo-Sang MJ, Lemij HG. Retinal nerve fiber layer measurements in normal black subjects as determined with scanning laser polarimetry. Ophthalmology. 1998 ; 105 : 78-81.
[21] Varma R, Wang D, Wu C, et al. Four-year incidence of open-angle glaucoma and ocular hypertension : the Los Angeles Latino Eye Study. Am J Ophthalmol. 2012 ; 154 : 315-25.
[22] Vitart V, Bencié G, Hayward C, et al. New loci associated with corneal central thickness include COL5A1, AKAP13 and AVGR8. Hum Mol Genet. 2010 ; 19 : 4304-11.
[23] Vithana EN, Aung T, Khor CC, et al. Collagen-related genes influence the glaucoma risk factor central corneal thickness. Hum Mol Genet. 2011 ; 20 : 649-58.
[24] Wang X, Harmon J, Zabrieskie N, et al. Using the Utah Population Database to assess familial risk of primary open angle glaucoma. Vision Res. 2010 ; 50 : 2391-5.
[25] Wiggs JL. The cell and molecular biology of complex forms of glaucoma : updates on genetic, environmental, and epigenetic risk factors. Invest Ophthalmol Vis Sci. 2012 ; 53 : 2467-9.
[26] Yap KL, Li S, Munoz-Cabello, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010 ; 38 : 662-74.
J.-P. Renard, H. El Chehab
Le rôle physiopathogénique de certains facteurs non héréditaires est mieux connu.
Un nombre croissant de données vient souligner les relations avec d’autres pathologies neurodégénératives liées à l’âge.
Le rôle des graisses alimentaires dans le GPAO est plus difficile à mettre en évidence chez l’homme.
À la lumière des nouvelles acquisitions des chapitres précédents, nous comprenons mieux l’implication de différents facteurs de risque (FDR) bien connus de la neuropathie optique glaucomateuse (NOG) et nous commençons à mieux cerner celle de nouveaux facteurs plus récemment rapportés en tant que FDR. L’objectif ici n’est pas d’aborder l’ensemble des FDR de la NOG, traités dans un autre chapitre, mais de tenter de préciser les particularités physiopathogéniques maintenant mieux connues de certains FDR non héréditaires, concernant essentiellement l’âge, ainsi que de nouveaux facteurs environnementaux liés au mode de vie. Enfin nous envisagerons le rôle physiopathogénique, direct ou non, de certains FDR oculaires indépendants des facteurs vasculaires et mécaniques déjà évoqués.
Toutes les études en accord avec ce FDR montrent que le risque de GPAO augmente progressivement tous les ans (OR entre 1,04 et 1,06 pour chaque année écoulée), et il n’y aurait pas d’âge « limite » au-delà duquel le risque augmente considérablement [18]. Nous avons vu dans la partie précédente de ce chapitre concernant les facteurs héréditaires le rôle rapporté des facteurs épigénétiques dans certaines maladies neurodégénératives chroniques comme la maladie d’Alzheimer, ainsi que dans les mécanismes physiopathologiques et la progression des maladies liées à l’âge [21, 28].
Ces données permettent de mieux comprendre l’augmentation de la prévalence du GPAO avec l’âge et d’en entrevoir les conséquences.
En France, 2 % des personnes âgées de plus de 40 ans seraient suivies pour un GPAO [5]. La méta-analyse réalisée sur 57 articles regroupant les résultats de 41 études séparées dont 34 randomisées avec un niveau de preuves élevé (plus de 86 000 participants) estime la prévalence globale du GPAO à 2,6 % (IC : 2,1-3,1). Elle varie en fonction de la tranche d’âge considérée [18, 24, 36] : de 1,3 % (IC : 0,9-1,9) pour des personnes entre 40 et 49 ans, à 5,1 % (IC : 3,6-7,2) pour les personnes entre 70 et 79 ans ; la prévalence est maximale au-delà de 80 ans où elle atteint 7,8 % (IC : 5,2-12). Après 70 ans, le risque de GPAO est multiplié par 12,2 par rapport aux sujets de moins de 50 ans [15, 23].
Des données plus récentes, en particulier concernant le dépistage de glaucomes non diagnostiqués lors de la mise en place de l’étude EMGT (Early Manifest Glaucoma Trial) chez un grand nombre de sujets (n = 32 918) âgés de 55 à 79 ans, retrouvent que la proportion de glaucomes dépistés augmente également avec l’âge. La prévalence des cas non connus détectés est cinq fois plus importante chez les patients âgés de 75 à 79 ans (2,73 %) par rapport aux patients de 55 à 59 ans (0,55 %) [23]. Ces résultats concernent à ce jour le plus important effectif de sujets caucasiens ayant bénéficié d’un dépistage du GPAO en Europe. Ils retrouvent et confirment la notion selon laquelle seulement 50 % des patients glaucomateux sont diagnostiqués [15].
Ainsi, la prévalence globale du GPAO de 2,6 % permet de retenir pour la France une estimation d’environ 1 700 000 patients atteints dont la moitié n’est pas diagnostiquée (65 800 000 habitants selon les données INSEE 2014). Si actuellement les sujets de 40 ans et plus (33 232 000) représentent 50,5 % de la population et ceux âgés d’au moins 65 ans 18 % (11 860 000), les estimations pour 2020 d’un tiers des Français âgés de plus de 65 ans laissent entrevoir dans un avenir proche un nombre important de patients liés à ce FDR.
À l’heure actuelle, peu de données concernent les facteurs environnementaux, nutritionnels ou liés au mode de vie.
Un certain nombre de FDR communs à ceux d’autres maladies neurodégénératives (maladie d’Alzheimer ou de Parkinson) ont été plus récemment évoqués parmi les multiples facteurs étiologiques du GPAO.
L’étude PHOTOGRAF réalisée en France, concernant l’évaluation des FDR connus et potentiels, différenciant les patients atteints de GPAO de ceux suivis pour une hypertension intra-oculaire (HTO), rapporte un certain nombre de données intéressantes. Réalisée à l’échelon national avec la collaboration d’épidémiologistes et de nutritionnistes, un de ses principaux objectifs a été d’évaluer plus spécifiquement les FDR associés aux modifications neurodégénératives dans le glaucome chez 339 GPAO et 339 HTO appariés selon l’âge (âge moyen : 63,5 ± 10 ans), le sexe et la durée de suivi de l’affection (76,4 ± 63 mois). Les résultats rapportent que l’exposition professionnelle aux pesticides est associée de façon significative au GPAO (OR : 2,65 ; IC : 1,04-6,78). La majorité concerne essentiellement des expositions à des insecticides, des herbicides et des fongicides. Dans cette étude, qui a fait l’objet de la première publication concernant ce type d’association – GPAO et pesticides –, la durée moyenne d’utilisation des pesticides durant la vie professionnelle des patients était de 23,9 ± 12,3 ans [31]. Une exposition professionnelle à d’autres produits chimiques n’a pas été retrouvée. Cette exposition professionnelle aux pesticides a été observée principalement chez les sujets travaillant dans le milieu agricole (fig. 8-12).
Un certain nombre d’études épidémiologiques retrouvent une association entre l’exposition professionnelle aux pesticides et un risque augmenté d’autres maladies neurodégénératives, comme la maladie d’Alzheimer [2, 34] ou de Parkinson [3, 12].
L’augmentation des maladies neurodégénératives cérébrales (maladie d’Alzheimer, maladie de Parkinson) a étroitement suivi l’utilisation croissante des pesticides agricoles durant les dernières décennies. Il est maintenant bien reconnu que les pesticides sont des FDR des maladies neurodégénératives cérébrales [4]. Une étude prospective bordelaise (étude PHYTONER) chez 614 vignerons (âge moyen environ 50 ans) rapporte que les sujets utilisant des pesticides depuis au moins 20 ans présentent une atteinte des fonctions cognitives et une diminution plus rapide du score MMS (Mini-Mental State) par rapport aux vignerons n’utilisant pas de pesticides. L’atteinte est plus importante chez les femmes, ainsi que chez les sujets à niveau intellectuel plus élevé et ne consommant pas d’alcool – les deux derniers étant des facteurs reconnus d’atteinte des fonctions cognitives ! Ces données soulignent que les vignerons exposés aux pesticides ont deux fois (OR : 1,97 ; IC : 1,09-3,59) plus de risque de perdre deux points au test du MMS en quatre à cinq ans par rapport aux vignerons non exposés [4]. L’association directe avec un type d’agent chimique spécifique n’a pas pu être identifiée à ce jour. Une étude américaine chez 3 084 agriculteurs de l’Utah montre que l’utilisation de pesticides augmente le risque de démence (OR : 1,38 ; IC : 1,09-1,76), et notamment le risque de maladie d’Alzheimer (OR : 1,42 ; IC : 1,06-1,96) [14]. Un lien avec d’autres maladies neurodégénératives a également été mis en évidence (dégénérescence frontotemporale, maladie de Parkinson, démences vasculaires, etc.) [4].
La toxicité des pesticides diffère en fonction du type d’agent chimique en cause. Elle pourrait être liée à un dysfonctionnement des mitochondries et de leur chaîne respiratoire pour les désinfectants par fumigation ou par une altération des synapses, ainsi que des neurotransmetteurs, pour les insecticides organophosphatés [39].
Les conséquences du dysfonctionnement mitochondrial sur les mécanismes pro-apoptotiques et apoptotiques des cellules ganglionnaires rétiniennes sont bien décrits dans le chapitre 8-I.
Comme pour la majorité des études concernant les pesticides, les patients atteints de GPAO dans l’étude française ne se souvenaient pas du type de pesticides utilisés, et l’association avec un agent chimique spécifique n’a pas été possible, d’autant plus que l’effectif des sujets concernés relativement limité (44 GPAO) n’aurait pas permis d’analyse en sous-groupes de pesticides [31].
Bien que le glaucome et les démences soient des pathologies neurodégénératives liées à l’âge, les preuves de leurs relations sont relativement récentes. L’étude de cohorte ALIENOR, réalisée en France sur 812 participants, indemnes de démence à l’inclusion, retrouve que les patients qui développaient une démence (41 patients) après trois ans de suivi présentaient statistiquement plus de GPAO (7/41 patients, soit 17 %) que les patients ne développant pas de démence (34/771 patients, soit 4 %). Ces sept patients glaucomateux ayant développé une démence avaient un glaucome à pression normale. Cette particularité, déjà notée dans des études précédentes, serait en faveur d’une physiopathologie commune pour les deux entités [16].
D’autres études se sont intéressées aux marqueurs de la maladie d’Alzheimer qui pourraient être présents dans les différents tissus oculaires. Ainsi il a été retrouvé des concentrations de la protéine TAU phosphorylée statistiquement plus élevées dans la rétine de patients glaucomateux ayant bénéficié d’une chirurgie filtrante par rapport à des témoins. Celle-ci se localise dans la couche nucléaire interne et plexiforme interne [17]. Un autre marqueur de la maladie d’Alzheimer, la protéine amyloïde β, est retrouvé en quantité diminuée dans le vitré de patients glaucomateux comme dans le liquide cérébrospinal (LCS) des patients atteints de démence [38]. Enfin, il a été rapporté le cas d’un patient glaucomateux, stable pendant plusieurs années (quatre ans), chez qui une progression rapide (six mois) apparaissait malgré le bon équilibre pressionnel. L’analyse du LCS a mis en évidence des anomalies des deux derniers biomarqueurs cités. Ces dernières sont connues pour être des facteurs d’aggravation de la maladie d’Alzheimer [27]. Pour terminer, très récemment plusieurs autres biomarqueurs de la maladie d’Alzheimer (ApoAL, ApoCIII, ApoE, TTR et α2M) ont été retrouvés à une forte concentration dans l’humeur aqueuse de patients atteints de GPAO [19].
En pratique, nous pouvons retenir qu’un nombre croissant de données vient souligner le lien qui semble exister entre ces deux maladies neurodégénératives, GPAO et maladie d’Alzheimer, dont la fréquence ne cesse d’augmenter avec en partage de nouveaux FDR, dont l’exposition aux agents neurotoxiques et en particulier l’exposition professionnelle qu’il faudra savoir rechercher chez tout patient glaucomateux.

Plusieurs études expérimentales ont évalué le rôle des acides gras poly-insaturés oméga 3 connus pour être impliqués dans l’activité des cellules rétiniennes, et notamment dans celle des cellules ganglionnaires atteintes dans le GPAO. Ces études réalisées chez l’animal (chien et rat) ont montré qu’un apport suffisant en acides gras poly-insaturés (omégas 3 et 6) permet une meilleure protection des cellules ganglionnaires rétiniennes par rapport à une carence d’un ou des deux acides gras [26, 30]. Un régime riche en oméga 3 réduit la pression intra-oculaire (PIO) en facilitant le flux d’humeur aqueuse par l’augmentation de la concentration en acide docosahexaénoïque dans le corps ciliaire chez le rat [30].
Le rôle des graisses alimentaires dans le GPAO est plus difficile à mettre en évidence chez l’homme. Les acides gras alimentaires influent sur les concentrations de prostaglandines endogènes (PGF2α) [20] ; la consommation de graisses alimentaires pourrait influer sur la PIO par l’intermédiaire des prostaglandines endogènes. Le rôle protecteur ou délétère des omégas 3, des omégas 6 et des acides gras mono- et poly-insaturés dans la survenue du glaucome est difficile à préciser chez l’homme en raison des régimes variés et du recueil des données. Si aucune différence significative sur le risque de GPAO n’a été mise en évidence entre les différents régimes (lipides totaux, graisses saturées, graisses mono- ou poly-insaturées), il semble que la consommation d’omégas 3 et 6 soit plus faible chez les patients atteints de GPAO [31].
L’étude PHOTOGRAF avait également pour objectif d’évaluer les associations potentielles du GPAO avec le mode de vie des patients, en particulier le régime alimentaire en acides gras oméga 3 ainsi que l’utilisation de compléments alimentaires, la consommation de tabac et d’alcool. Un questionnaire détaillé élaboré avec la collaboration de nutritionnistes a permis de préciser la fréquence de la consommation d’aliments riches en omégas 3, en particulier celles de poissons (blanc, gras), de noix (précurseurs de l’acide linoléique et α-linolénique – omégas 3 ayant une action anti-inflammatoire et régulatrice du cholestérol) et d’huiles, chez les 339 patients ayant un GPAO, et de la comparer avec celle relevée au sein du groupe témoin de 339 patients ayant une HTO isolée, appariés selon l’âge, le sexe et l’ancienneté de l’affection.
Le questionnaire spécifiquement développé pour l’étude comportait des questions sur l’usage quantitatif et préférentiel de certaines huiles et matières grasses alors classées en : riches en oméga 3 (huiles de colza, de noix et de soja), riches en oméga 6 (huile de tournesol, de maïs et de pépins de raisin) et riches en acides gras mono-insaturés (huile d’olives). Pour chaque type d’huile, trois groupes de consommateurs étaient définis : consommation importante, intermédiaire ou rare (fig. 8-13).
La plus faible consommation de poissons gras (inférieure à une fois par semaine) chez les patients atteints de GPAO, suivis en moyenne depuis sept ans, était significative (OR : 2,14 ; IC95% : 1,10-4,17), ainsi que la plus faible consommation de noix (mois de huit fois par an) (OR : 2,02 ; IC95% : 1,18-3,47). La consommation des différents autres types d’huile (oméga 6) et de poissons blancs ne montrait pas de différence significative avec le groupe témoin suivi pour HTO [31].
Ces résultats suggèrent un rôle protecteur potentiel des acides gras poly-insaturés oméga 3 dans le GPAO qui pourrait être expliqué par leur effet vasculaire et neuroprotecteur. Le rôle préventif des maladies cardiovasculaires est bien établi avec un effet sur la fonction endothéliale [22], qui peut aussi être impliqué dans l’étiologie du glaucome [33]. De fortes concentrations plasmatiques et un régime riche en oméga 3 ont été associés avec une réduction du risque de maladie d’Alzheimer et pourraient partager un mécanisme commun avec le glaucome [7, 37]. Les omégas 3 ont également une fonction structurale et protectrice dans la rétine et ont été associés avec un risque réduit de dégénérescence maculaire liée à l’âge [8, 25].
En fait, c’est le rapport oméga 6/oméga 3 qui semble le plus important et qui, selon les recommandations de l’AFSSA (Agence française de sécurité sanitaire des aliments), devrait être proche de cinq, c’est-à-dire que l’alimentation devrait apporter cinq molécules d’oméga 6 pour une d’oméga 3, mais ce rapport est souvent plus élevé (favorisant l’obésité). Des études sont encore nécessaires pour une meilleure évaluation de la consommation des différents types d’acides gras.
On peut retenir qu’il semblerait qu’un apport nutritif suffisant en acides gras poly-insaturés de type oméga 3 permettrait de diminuer l’incidence du GPAO. Ces données sont en accord avec l’effet protecteur des acides gras pour les maladies cardiovasculaires [22] et neurodégénératives [1] avec lesquelles le glaucome partage des similitudes physiopathogéniques [31].

Fig. 8-13 Aliments riches en oméga 3.
Différents marqueurs du polymorphisme génique, comme nous l’avons vu dans le chapitre précédent, ont montré leur implication dans les variations de certains paramètres cliniques comme l’épaisseur cornéenne centrale [11, 35].
Des études récentes rapportent des associations entre l’épaisseur cornéenne centrale et différents loci de gènes (gènes COL5A1, COL8A2, FOXO1, AKAP13, etc.). Ces gènes impliqués dans le métabolisme du collagène stromal cornéen ne sont pas directement liés à une augmentation de la prévalence de glaucome [13]. Seule la mutation du gène COL8A2 aurait une implication, et cette mutation a été observée dans un groupe de patients glaucomateux caucasiens ayant une pachymétrie faible [9].
L’épaisseur cornéenne centrale doit plus vraisemblablement être considérée comme FDR indirect à cause de son influence sur la mesure de la PIO, mais le débat concernant son rôle potentiel en tant que FDR de GPAO n’a pas sa place dans ce chapitre.
La myopie est un facteur risque du GPAO indépendant des autres facteurs [6].
Sur le plan physiopathologique, l’anatomie et la morphologie de la tête du nerf optique (TNO) du sujet myope le rendraient plus susceptible aux dommages glaucomateux que le sujet emmétrope. L’insertion oblique de la TNO est à l’origine de forces de cisaillement plus importantes. Les patients myopes sont caractérisés par l’existence d’une lame criblée au niveau de la TNO bien plus fine. On comprend ainsi aisément que pour un niveau donné de PIO, le gradient de pression translaminaire soit plus important avec une plus grande vulnérabilité des fibres nerveuses rétiniennes de la TNO, facilitant ainsi l’apparition et la progression du GPAO. Par ailleurs, l’existence d’une élongation axiale et d’un remodelage scléral est à l’origine d’une diminution de la résistance à l’expansion sclérale pour un niveau donné de PIO. Enfin, les facteurs vasculaires jouent un rôle important. Une diminution du flux sanguin au niveau des artères ciliaires courtes postérieures chez les sujets myopes, qui augmente avec l’élévation de la réfraction myopique, peut ainsi expliquer que la progression du glaucome chez les myopes forts soit également en rapport avec une diminution de la perfusion de la TNO. Ainsi la sensibilité particulière de l’œil myope à l’HTO, liée et en relation avec les forces de traction au niveau de la lame criblée, fait de la myopie un FDR indépendant de survenue et de développement du glaucome. Cependant, l’atteinte glaucomateuse n’est pas proportionnelle au degré de myopie ni à l’anisométropie myopique entre les deux yeux [10].
Le rôle physiopathogénique du flux sanguin oculaire et la pression de perfusion oculaire moyenne ont été largement abordés dans les chapitres précédents. L’hypothèse actuellement retenue est celle de l’existence d’un rôle majeur des fluctuations de la pression artérielle et de la pression de perfusion oculaire liées à un trouble de la régulation vasculaire dans les mécanismes pathogéniques de la NOG [29, 32].
Les connaissances physiopathogéniques de la neuropathie optique glaucomateuse ont donc nettement progressé. Les facteurs épigénétiques permettent de mieux appréhender et comprendre l’augmentation de l’incidence du GPAO avec l’âge. Surtout, une meilleure connaissance des affections neurodégénératives nous a permis de progresser dans l’approche de certains mécanismes neurotoxiques communs, liés au mode de vie courant. La mise en évidence de nouveaux FDR environnementaux nécessite qu’ils doivent désormais être considérés avec attention pour une prise en charge adaptée des patients atteints de GPAO.
Retenir
Un nombre croissant de données souligne le lien qui semble exister entre les maladies neurodégénératives, GPAO et maladie d’Alzheimer.
Le rôle physiopathogénique de l’exposition neurotoxique rapporté doit inciter la recherche systématique d’une exposition à ces agents chez tout patient glaucomateux.
Des études sont encore nécessaires pour une meilleure évaluation du rôle de la consommation des différents types d’acides gras.
[1] Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J Clin Nutr. 2006 ; 83(Suppl. 6) : 1467S-1476S.
[2] Baldi I, Cantagrel A, Lebailly P, et al. Association between Parkinson’s disease and exposure to pesticides in southwestern France. Neuroepidemiology. 2003 ; 22 : 305-10.
[3] Baldi I, Lebailly P, Mohammed-Brahim B, et al. Neurodegenerative diseases and exposure to pesticides in the elderly. Am J Epidemiol. 2003 ; 157 : 409-14.
[4] Baldi I, Gruber A, Rondeau V, et al. Neurobehavioral effects of long-term exposure to pesticides : results from the 4-year follow-up of the PHYTONER study. Occup Environ Med. 2011 ; 68 : 108-15.
[5] Bron A, Chaine G, Villain M, et al. [Risk factors for primary open-angle glaucoma]. J Fr Ophtalmol. 2008 ; 31 : 435-44.
[6] Chang RT, Singh K. Myopia and glaucoma : diagnostic and therapeutic challenges. Curr Opin Ophthalmol. 2013 ; 24 : 96-101.
[7] Cunnane SC, Plourde M, Pifferi F, et al. Fish, docosahexaenoic acid and Alzheimer’s disease. Prog Lipid Res. 2009 ; 48 : 239-56.
[8] Delcourt C, Carriere I, Cristol JP, et al. Dietary fat and the risk of age-related maculopathy : the POLANUT study. Eur J Clin Nutr. 2007 ; 61 : 1341-4.
[9] Desronvil T, Logan-Wyatt D, Abdrabou W, et al. Distribution of COL8A2 and COL8A1 gene variants in Caucasian primary open angle glaucoma patients with thin central corneal thickness. Mol Vis. 2010 ; 16 : 2185-91.
[10] Detry-Morel M. [Is myopia a risk factor for glaucoma ?]. J Fr Ophtalmol. 2011 ; 34 : 392-5.
[11] Dimasi DP, Burdon KP, Hewitt AW, et al. Genetic investigation into the endophenotypic status of central corneal thickness and optic disc parameters in relation to open-angle glaucoma. Am J Ophthalmol. 2012 ; 154 : 833-42.
[12] Elbaz A, Tranchant C. Epidemiologic studies of environmental exposures in Parkinson’s disease. J Neurol Sci. 2007 ; 262 : 37-44.
[13] Gao X, Gauderman WJ, Liu Y, et al. A genome-wide association study of central corneal thickness in Latinos. Invest Ophthalmol Vis Sci. 2013 ; 54 : 2435-43.
[14] Hayden KM, Norton MC, Darcey D, et al. Occupational exposure to pesticides increases the risk of incident AD : the Cash County study. Neurology. 2010 ; 74 : 1524-30.
[15] Heijl A, Bengtsson B, Oskarsdottir SE. Prevalence and severity of undetected manifest glaucoma : results from the Early Manifest Glaucoma Trial Screening. Ophthalmology. 2013 ; 120 : 1541-5.
[16] Helmer C, Malet F, Rougier MB, et al. Is there a link between open-angle glaucoma and dementia ? The 3C-Alienor Cohort. Ann Neurol. 2013 (sous presse).
[17] Ho WL, Leung Y, Tsang AWT, et al. Review : Tauopathy in the retina and optic nerve : does it shadow pathological changes in the brain ? Mol Vis. 2012 ; 18 : 2700-10.
[18] Hollands H, Johnson D, Hollands S, et al. Do findings on routine examination identify patients at risk for primary open-angle glaucoma ? The rational clinical examination systematic review. JAMA. 2013 ; 309 : 2035-42.
[19] Inoue T, Kawaji T, Tanihara H. Elevated levels of multiple biomarkers of Alzheimer’s disease in the aqueous humor of eyes with open angle glaucoma. Invest Ophthalmol Visc Sci. 2013 ; 54 : 5353-8.
[20] Kang JH, Pasquale LR, Willett WC, et al. Dietary fat consumption and primary open-angle glaucoma. Am J Clin Nutr. 2004 ; 79 : 755-64.
[21] Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012 ; 81 : 303-11.
[22] Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002 ; 106 : 2747-57.
[23] Le A, Mukesh BN, McCarty CA, Taylor HR. Risk factors associated with the incidence of open-angle glaucoma : the visual impairment project. Invest Ophthalmol Vis Sci. 2003 ; 44 : 3783-9.
[24] Leske MC, Wu SY, Hennis A, et al. BESs Study Group. Risk factors for incident open-angle glaucoma : the Barbados Eye Studies. Ophthalmology. 2008 ; 115 : 85-93.
[25] Merle B, Delyfer MN, Korobelnik JF, et al. Dietary omega-3 fatty acids and the risk for age-related maculopathy : the Alienor study. Invest Ophthalmol Visc Sci. 2011 ; 52 : 6004-11.
[26] Nguyen CTO, Vingrys AJ, Bui BV. Dietary ω-3 deficiency and IOP insult are additive risk factors for ganglion cell dysfunction. J Glaucoma. 2013 ; 22 : 269-77.
[27] Nucci C, Martucci A, Martorana A, et al. Glaucoma progression associated with altered cerebral spinal fluid levels of amyloid beta and tau proteins. Clin Experiment Ophthalmol. 2011 ; 39 : 279-81.
[28] Qureshi IA, Mehler MF. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep. 2011 ; 11 : 464-73.Tuck MW, Crick RP. The age distribution of primary open angle glaucoma. Ophthalmic Epidemiol. 1998 ; 5 : 173-83.
[29] Ramdas WD, Wolfs RCW, Hofman A, et al. Ocular perfusion pressure and the incidence of glaucoma : real effect or artifact ? The Rotterdam Study. Invest Ophthalmol Vis Sci. 2011 ; 52 : 6875-81.
[30] Ren H, Magulike N, Ghebremeskel K, Crawford M. Primary open-angle glaucoma patients have reduced levels of blood docosahexaenoic and eicosapentaenoic acids. Prostaglandins Leukot Essent Fatty Acids. 2006 ; 74 : 157-63.
[31] Renard JP, Rouland JF, Bron A, et al. Nutritional, lifestyle and environmental factors in ocular hypertension and primary open-angle glaucoma : an exploratory case-control study. Acta Ophthalmol. 2013 ; 91 : 505-13.
[32] Renard JP. [High blood pressure-low ocular pressure : the parallels. Vascular risk factors in glaucoma]. J Fr Ophtalmol. 2007 ; 30 (5 Pt 2) : 3S18-21.
[33] Resh H, Garhofer G, Fuchsjager-Mayrl G, et al. Endothelial dysfunction in glaucoma Acta Ophtalmol. 2009 ; 87 : 4-12.
[34] Santibanez M, Bolumar F, Garcia AM. Occupational risk factors in Alzheimer’s disease : a review assessing the quality of published epidemiological studies. Occup Environ Med. 2007 ; 64 : 723-32.
[35] Scheetz TE, Fingert JH, Wang K, et al. A genome-wide association study for primary open angle glaucoma and macular degeneration reveals novel loci. Plos One. 2013 ; 8 : e58657.
[36] Tuck MW, Crick RP. The age distribution of primary open angle glaucoma. Ophthalmic Epidemiol. 1998 ; 5 : 173-83.
[37] Wostyn MR, Audenaert K, De Deyn PP. Alzheimer’s disease and glaucoma is there a causal relationship ? Br J Ophthalmol. 2009 ; 93 : 1557-59.
[38] Yoneda S, Hara H, Hirata A, et al. Vitreous fluid levels of beta-amyloid (1-42) and tau in patients with retinal diseases. Jpn J Ophthalmol. 2005 ; 49 : 106-8.
[39] Zaganas I, Kapetanaki S, Mastorodemos V, et al. Linking pesticide exposure and dementia : what is the evidence ? Toxicology. 2013 ; 307 : 3-11.
A. Bron
Les cellules ganglionnaires de la rétine et leurs dysfonctionnements sont à la base de la neuropathie optique glaucomateuse.
Avant de mourir, ces cellules présentent des étapes intermédiaires ou les fonctions qu’elles doivent normalement assurer sont perturbées.
Ces altérations ne sont pas homogènes suivant la partie du neurone considéré ; on parle de compartimentalisation.
Les glaucomes, quel que soit leur type, partagent un substratum anatomique commun, la perte des cellules ganglionnaires supérieure à celle liée à l’âge. Ces cellules ganglionnaires sont en fait diverses et il est dénombré plus de 15 types différents chez les mammifères [22]. Leurs différences sont anatomiques mais aussi physiologiques, et elles codent pour des informations différentes. Un aspect fascinant récemment souligné est la possibilité qu’ont certaines cellules ganglionnaires, dont le nombre est estimé à 0,2 % du contingent total, d’utiliser un pigment capable de phototransduction, la mélanopsine [22]. Ainsi ces cellules ganglionnaires particulières sont-elles impliquées dans les phénomènes liés à la lumière et particulièrement le rythme circadien.
Lors des glaucomes, le site majeur de la souffrance de ces cellules ganglionnaires est la lame criblée, comme l’ont bien démontré Quigley et Addicks il y a environ 30 ans [20]. Les travaux modernes sur la lame criblée ont confirmé ces données. Cependant, confiner la physiopathogénie des glaucomes aux seules cellules ganglionnaires est un peu réducteur et plusieurs auteurs ont écorné ce dogme. Comme le soulignent Calkins et Horner [5], ce qui est le plus destructeur dans les glaucomes ne se passe pas dans l’œil mais plutôt à l’extérieur de l’œil, le long des voies visuelles et dans les zones cérébrales connectées aux afférences visuelles. C’est un peu à la lueur de ces nouvelles interrogations que nous évoquerons ici les trois points suivants :
la compartimentalisation des neurones ;
l’atteinte en amont des cellules ganglionnaires ;
l’atteinte en aval des cellules ganglionnaires.
Un neurone est constitué de trois éléments distincts mais néanmoins connectés entre eux : l’arbre dendritique, le corps cellulaire ou soma et l’axone. Les cellules ganglionnaires ont un arbre dendritique très développé, une cellule possédant à elle seule environ 10 000 synapses. Leur axone est particulièrement long et, si l’on attribue au corps cellulaire la taille d’une pomme, la distance du corps cellulaire au corps genouillé latéral où cet axone fait relais chez les primates humains et non humains est d’environ 800 mètres. Il est donc facile de concevoir que ce qui se passe à un endroit d’un neurone aussi étendu peut être tout à fait localisé avant d’impacter la survie du neurone globalement [28].
Des processus d’autodestruction ont été identifiés au niveau de l’axone, des dendrites et des synapes [28]. Mais avant de mourir, plusieurs étapes ont été décrites, notamment le dysfonctionnement des dendrites et le réarrangement synaptique. Il existe des preuves expérimentales que le premier élément qui survient n’est pas une perte tissulaire mais un dysfonctionnement de la physiologie du neurone [10]. Cette anomalie est au mieux captée par l’électrophysiologie mais on connaît les difficultés à extraire un signal pertinent, du bruit de fond. Dès lors, la fonction est altérée avant même la perte cellulaire [28].
Cela explique aussi que ce que l’on peut analyser avec les imageurs aujourd’hui ne reflète pas forcément la physiologie. Avant d’avoir une perte en fibre sur un OCT, par exemple, l’épaisseur des fibres peut être encore normale alors que les neurones dysfonctionnent. De plus, un delta constant de 15 % sur un modèle expérimental de glaucome chez le singe semblerait indiquer que la perte des cellules ganglionnaires observée en péripapillaire n’est pas la même que celle réellement présente au niveau du nerf optique en rétrolaminaire [8].
Dans un article récent rapportant l’évolution de la mort des cellules ganglionnaires après une compression du nerf optique chez la souris, il a bien été montré que ce sont d’abord les dendrites qui souffrent sous la forme anatomique d’une contraction du champ dendritique et de la diminution de la richesse synaptique [14]. Dans ce travail, les cellules ganglionnaires possédant un champ dendritique plus large au départ survivaient mieux que celles avec un champ plus étroit et montraient une contraction dendritique plus modérée. Puis l’axone était menacé à son tour et enfin le corps cellulaire des cellules ganglionnaires.
Déjà en 1998 Weber et al. [26] avaient montré un amincissement des dendrites et une diminution de la complexité de l’arbre dendritique sur un modèle de glaucome chez le singe. Ils avaient observé que ce phénomène touchait à la fois les cellules de la voie magno- et parvocellulaire, mais ils notaient à l’époque que les anomalies dendritiques étaient plus marquées pour les cellules de la voie magnocellulaire. Ce phénomène est constant quelle que soit l’espèce étudiée – rongeurs, félins ou primates. Il existe aussi bien dans les modèles expérimentaux de glaucomes que chez l’homme glaucomateux [15]. On parle également de « pruning », qui se traduit littéralement par élagage et qui témoigne de l’appauvrissement de l’arbre dendritique et donc du nombre des connexions [4]. Dès lors, on saisit mieux l’intérêt de l’étude de la couche plexiforme interne où les dendrites des cellules ganglionnaires s’articulent avec les axones des cellules bipolaires, horizontales et amacrines. C’est malheureusement pour l’instant une couche de la rétine assez difficile à isoler et délimiter de la couche des corps cellulaires des cellules ganglionnaires avec nos moyens d’imagerie tels que l’OCT.
Ces modifications dendritiques sont réparties sur toute la voie visuelle. Elles ont été mises en évidence chez le singe au niveau du corps genouillé latéral lors d’un glaucome induit par laser [13]. Ces modifications dendritiques ne sont pas propres aux glaucomes mais ont également été observées dans des maladies neurodégénératives comme la maladie d’Alzheimer et la sclérose latérale amyotrophique [15]. Il s’agit principalement d’une contraction et d’une raréfaction de ces dendrites et du nombre de leurs connexions. Ce phénomène est également connu dans l’ischémie cérébrale. C’est en outre un constat réalisé lors du développement cérébral où il existe un profond réarrangement des dendrites au niveau des cellules ganglionnaires mais aussi au niveau du cortex visuel.
Les facteurs de croissance déjà cités tels que le BDNF (brain-derived neurotrophic factor), le NGF (nerve growth factor) et d’autres neurotrophines contrôleraient ces modifications et les connexions des neurones corticaux [1]. Pendant le développement du système nerveux, les neurones ont besoin de ces facteurs de croissance pour leur survie mais également pour établir des connexions avec les autres neurones via les synapses [28].
Les glaucomes partagent des points communs avec d’autres maladies neurodégénératives telles que la maladie d’Alzheimer, la maladie de Parkinson ou la sclérose latérale amyotrophique [16]. Une similarité mécanistique de ces maladies est la neuro-inflammation et plusieurs travaux ont souligné les liens entre la cascade du complément et des modèles expérimentaux de glaucomes [16]. L’activation du complément, notamment par les astrocytes activés, pourrait participer à ces anomalies dendritiques et à ce mécanisme de raréfaction synaptique [1].
Enfin, l’âge modifie également cet arbre dendritique en le raréfiant et en l’atrophiant aussi bien chez l’homme que chez l’animal.
Comment concevoir qu’un processus physiologique touche des cellules spécifiques à un endroit donné et qu’à 200 µm de distance les autres cellules ne soient pas touchées ? C’est un dogme qui a soulevé des controverses il y a une quinzaine d’années, notamment avec Jost Jonas qui a osé mettre en doute le fait que les photorécepteurs n’étaient pas atteints dans les glaucomes [18]. De façon élégante, il a montré que des pertes en photorécepteurs pouvaient être observées chez des glaucomateux. Las, Quigley est venu tout remettre dans l’ordre établi par un éditorial un peu cinglant [19]. Cependant, l’arrivée de l’optique adaptative a ranimé ces interrogations et effectivement il existe des publications montrant qu’avec cette technique il est possible de mettre en évidence des altérations des photorécepteurs [7, 27]. En effet, Werner et al. ont montré en OCT que la longueur de l’article externe des photorécepteurs est diminuée chez des patients glaucomateux et particulièrement aux endroits correspondants aux scotomes mis en évidence sur le champ visuel. Des anomalies similaires ont été observées sur des modèles animaux de glaucome avec également une diminution de la densité des photorécepteurs [11]. Cette idée n’est pas aussi saugrenue qu’il y paraît car les cônes et les cellules ganglionnaires partagent en commun le même pigment, la mélanopsine [3].
Le flux orthograde permet aux neurones de véhiculer une information essentiellement sous la forme d’un potentiel d’action transmembranaire dans le sens dendrite-soma-axone. Il existe un flux rétrograde tout aussi important qui transmet des facteurs de croissance dans le sens inverse. Ces facteurs de survie sont essentiels pour la survie du neurone. L’interruption de ce flux conduit à la mort neuronale. Le passage de la lame criblée pour les axones des cellules ganglionnaires représente un défi physiologique car c’est à ce niveau que sont concentrées toutes les contraintes biomécaniques de l’œil [2]. Les axones seraient donc alors soumis à un processus d’autodestruction principalement piloté par l’apoptose mais pas uniquement. D’autres phénomènes comme le système ubiquitine protéasome, indépendant des caspases, serait impliqué [28]. Whitmore et al. [28] décrivent deux grands types d’atteinte des axones qui sont par ailleurs bien connus dans les maladies neurodégénératives du système nerveux central mais également dans les neuropathies périphériques :
la dégénérescence wallérienne antérograde qui surviendrait plus volontiers dans les traumatismes axonaux importants ;
le « dying-back » qui, lui, propage les signaux de mort au soma de façon rétrograde et correspondrait à des lésions plus chroniques [28].
Lors d’un modèle expérimental de glaucome chez le singe, il a été montré une diminution de l’épaisseur des couches au niveau du corps genouillé latéral, aussi bien pour les couches ventrales recevant les terminaisons des cellules ganglionnaires de la voie magnocellulaire que dans la région dorsale où est localisée la voie parvocellulaire [29]. La voie koniocellulaire est également altérée [30]. Dans un modèle identique, il a cependant été montré que les cellules de la voie magnocellulaire sont plus fortement impactées que celles de la voie parvocellulaire au niveau du corps genouillé latéral [28]. Il s’agit d’une controverse ancienne qui a ses partisans pour chaque hypothèse [17, 21]. Cependant, il semble qu’en clinique les choses ne soient pas si tranchées et que toutes les voies soient affectées ou que du moins nos moyens de détection ne soient pas aussi spécifiques que l’on pouvait l’espérer. Ainsi, les espoirs qui avaient été mis dans la périmétrie bleu jaune pour détecter des anomalies plus précoces pourraient être déçus par une publication récente rapportant que les déficits du champ visuel ne semblaient pas plus précoces qu’en périmétrie blanc/blanc qui caractérise la voie parvocellulaire [23]. Ces signes de souffrance neuronale sont également retrouvés mais à une moindre échelle sur les projections du corps genouillé latéral et du cortex de l’œil qui n’a pas subi d’élévation de la pression intra-oculaire (PIO) [30]. Cela soulève l’aspect intrigant du caractère bilatéral et asymétrique de la présentation des glaucomes ainsi que de la « transmission » de la maladie à l’œil non atteint soit par voie humorale, soit par voie de contiguïté, la glie par exemple [9]. Cette notion de signal transmissible est bien mise en évidence dans les modèles animaux de glaucome ou du moins d’élévation de la PIO. Ainsi, en augmentant la PIO d’un côté par le laser, on augmente dans cet œil l’activation de la glie bien mise en évidence par la coloration de la GFAP (glial fibrillary acid protein). Toutefois, sur l’œil controlatéral qui n’a pas eu d’induction d’augmentation de la PIO, on retrouve également une augmentation de l’activation de la glie, certes plus modérée, donnant à ce type de glaucome un aspect sympathique comme il a été décrit pour l’ophtalmie sympathique (fig. 8-14) [9]. Dès lors, Vidal Sanz et al. [24] mettent en garde contre l’utilisation de cet œil controlatéral comme contrôle car la glie est également activée. L’atteinte des voies optiques se complète au niveau du cortex occipital et Yucel a bien montré en histologie une diminution de l’épaisseur des couches corticales chez l’homme et au niveau du corps genouillé latéral [12]. Ces remaniements à type de diminution de taille des neurones, de rétrécissement, existent également dans ce même modèle au niveau du cortex occipital [30]. Ainsi cette mort cellulaire se transmet tout au long du tractus visuel.
Ces constatations sont intéressantes car elles soulèvent la question d’un signal transmissible de part en part de la voie visuelle principale à la fois ipsilatéral mais également controlatéral. Elles soulignent également la difficulté d’une réhabilitation visuelle dans les pathologies glaucomateuses. Ainsi, s’il est possible expérimentalement de refaire fonctionner des photorécepteurs dans la rétinopathie pigmentaire par exemple, l’intégrité du reste de la voie visuelle est nécessaire pour atteindre le cortex visuel [3].

Fig. 8-14 a. Coupe rétinienne sur un œil de rat sans aucune intervention. Les noyaux cellulaires figurent en bleu (coloration DAPI – di-aminido-phényl-indol). b. Coupe rétinienne sur un œil de rat trois jours après une élévation de la pression intra-oculaire par laser. L’activation gliale est marquée par la coloration GFAP. c. Coupe rétinienne sur l’œil controlatéral du même animal. Cet œil n’a pas reçu de traitement par laser mais il existe une activation de la glie a minima et particulièrement sur les couches internes de la rétine.
GCL : couche des cellules ganglionnaires ; INL : couche des cellules bipolaires ; ONL : couche des photorécepteurs. (D’après Fourgeux et al., 2012 [9].)
La préoccupation thérapeutique essentielle pour les glaucomes en 2014 demeure l’abaissement de la PIO. Néanmoins, l’« ouverture » de la neuropathie optique glaucomateuse aux règles qui prévalent pour les neurones est passionnante car elle autorise des espoirs d’approches thérapeutiques nouvelles comme la neuroprotection, la neurorégénération ou le renforcement neuronal par exemple [6].
Retenir
La glie, tissu non neuronal, joue un rôle majeur d’information, de support métabolique et de couplage entre vaisseaux et neurones.
Chaque constituant de la cellule ganglionnaire est important à considérer (dendrites, corps cellulaire, axone).
Néanmoins, c’est le réarrangement synaptique et la richesse de l’arbre synaptique (environ 10 000 connexions pour une cellule ganglionnaire) qui donne son « intelligence » au neurone et la base pour la plasticité neuronale.
[1] Almasieh M, Wilson AM, Morquette B, et al. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retinal Eye Res. 2012 ; 31 : 152-81.
[2] Burgoyne CF. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exper Eye Res. 2010 ; 93 : 120-32.
[3] Busskamp V, Duebel J, Balya D, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010 ; 329 : 413-7.
[4] Calkins DJ. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retinal Eye Res. 2012 ; 31 : 702-19.
[5] Calkins DJ, Horner PJ. The cell and molecular biology of glaucoma : axonopathy and the brain. Invest Ophthalmol Vis Sci. 2012 ; 53 : 2482-4.
[6] Chang EE, Goldberg JL. Glaucoma 2.0 : neuroprotection, neuroregeneration, neuroenhancement. Ophthalmology. 2012 ; 119 : 979-86.
[7] Choi SS, Zawadzki RJ, Lim MC, et al. Evidence of outer retinal changes in glaucoma patients as revealed by ultrahigh-resolution in vivo retinal imaging. Br J Ophthalmol. 2011 ; 95 : 131-141.
[8] Cull GA, Reynaud J, Wang L, et al. Relationship between orbital optic nerve axon counts and retinal nerve fiber layer thickness measured by spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2012 ; 53 : 7766-73.
[9] Fourgeux C, Martine L, Pasquis B, et al. Steady-state levels of retinal 24S-hydroxycholesterol are maintained by glial cells intervention after elevation of intraocular pressure in the rat. Acta Ophthalmol. 2012 ; 90 : 560-7.
[10] Frankfort BJ, Khan AK, Tse DY, et al. Elevated intraocular pressure causes inner retinal dysfunction prior to cell loss in a mouse model of experimental glaucoma. Invest Ophthalmol Vis Sci. 2013 ; 54 : 762-70.
[11] Guo L, Normando EM, Nizari S, et al. Tracking longitudinal retinal changes in experimental ocular hypertension using the cSLO and spectral domain-OCT. Invest Ophthalmol Vis Sci. 2010 ; 51 : 6504-13.
[12] Gupta N, Greenberg G, de Tilly LN, et al. Atrophy of the lateral geniculate nucleus in human glaucoma detected by magnetic resonance imaging. Br J Ophthalmol. 2009 ; 93 : 56-60.
[13] Gupta N, Ly T, Zhang Q, et al. Chronic ocular hypertension induces dendrite pathology in the lateral geniculate nucleus of the brain. Exper Eye Res. 2007 ; 84 : 176-184.
[14] Leung CK, Weinreb RN, Li ZW, et al. Long-term in vivo imaging and measurement of dendritic shrinkage of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2011 ; 52 : 1539-47.
[15] Liu M, Duggan J, Salt TE, Cordeiro MF. Dendritic changes in visual pathways in glaucoma and other neurodegenerative conditions. Exper Eye Res. 2011 ; 92 : 244-250.
[16] McKinnon SJ. The cell and molecular biology of glaucoma : common neurodegenerative pathways and relevance to glaucoma. Invest Ophthalmol Vis Sci. 2012 ; 53 : 2485-2487.
[17] Morgan JE. Retinal ganglion cell shrinkage in glaucoma. J Glaucoma. 2002 ; 11 : 365-70.
[18] Panda S, Jonas JB. Decreased photoreceptor count in human eyes with secondary angle-closure glaucoma. Invest Ophthalmol Vis Sci. 1992 ; 33 : 2532-6.
[19] Quigley HA. Selective citation of evidence regarding photoreceptor loss in glaucoma. Arch Ophthalmol. 2001 ; 119 : 1390-1.
[20] Quigley HA, Addicks EM. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch Ophthalmol. 1981 ; 99 : 137-43.
[21] Quigley HA, Sanchez RM, Dunkelberger GR, et al. Chronic glaucoma selectively damages large optic nerve fibers. Invest Ophthalmol Vis Sci. 1987 ; 28 : 913-20.
[22] Sand A, Schmidt TM, Kofuji P. Diverse types of ganglion cell photoreceptors in the mammalian retina. Prog Retinal Eye Res. 2012 ; 31 : 287-302.
[23] van der Schoot J, Reus NJ, Colen TP, Lemij HG. The ability of short-wavelength automated perimetry to predict conversion to glaucoma. Ophthalmology. 2010 ; 117 : 30-4.
[24] Vidal-Sanz M, Salinas-Navarro M, Nadal-Nicolas FM, et al. Understanding glaucomatous damage : anatomical and functional data from ocular hypertensive rodent retinas. Prog Retinal Eye Res. 2011 ; 31 : 1-27.
[25] Weber AJ, Chen H, Hubbard WC, Kaufman PL. Experimental glaucoma and cell size, density, and number in the primate lateral geniculate nucleus. Invest Ophthalmol Vis Sci. 2000 ; 41 : 1370-9.
[26] Weber AJ, Kaufman PL, Hubbard WC. Morphology of single ganglion cells in the glaucomatous primate retina. Invest Ophthalmol Vis Sci. 1998 ; 39 : 2304-20.
[27] Werner JS, Keltner JL, Zawadzki RJ, Choi SS. Outer retinal abnormalities associated with inner retinal pathology in nonglaucomatous and glaucomatous optic neuropathies. Eye (Lond.). 2011 ; 25 : 279-89.
[28] Whitmore AV, Libby RT, John SW. Glaucoma : thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes ? Prog Retinal Eye Res. 2005 ; 24 : 639-62.
[29] Yucel YH, Zhang Q, Gupta N, et al. Loss of neurons in magnocellular and parvocellular layers of the lateral geniculate nucleus in glaucoma. Arch Ophthalmol. 2000 ; 118 : 378-84.
[30] Yucel YH, Zhang Q, Weinreb RN, et al. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Prog Retinal Eye Res. 2003 ; 22 : 465-81
- Chapitre 8
Physiopathogénie de la neuropathie optique glaucomateuse- I - Dégénérescence neurorétinienne glaucomateuse
- II - Facteurs anatomiques et théorie biomécanique de la neuropathie optique glaucomateuse
- III - Facteurs vasculaires de la neuropathie optique glaucomateuse
- IV - Prédisposition génétique et autres facteurs
- V - Physiopathogénie de la neuropathie optique glaucomateuse : nouvelles pistes