Physiopathologie générale des œdèmes maculaires
Coordonné par P. Massin, R. Tadayoni
R. Tadayoni
L’œdème maculaire est classiquement diagnostiqué et même défini par un épaississement maculaire dont la cause est une rupture de la barrière hémato-rétinienne (BHR) et la conséquence une baisse d’acuité visuelle. En effet, en conditions normales, un système complexe permet de conserver des conditions optimales pour le fonctionnement de la rétine externe et interne malgré la présence d’un environnement liquidien (vitré), d’un haut niveau d’énergie (lumière), de forts niveaux de transfert d’oxygène (choriocapillaire) et de la nécessité d’un milieu extracellulaire parfaitement contrôlé et détoxifié pour la neurotransmission.
Ce système complexe inclut le contrôle précis des échanges avec le sang des vaisseaux rétiniens (barrière hémato-rétinienne interne ou BHRi) et de la choriocapillaire (barrière hématorétinienne externe ou BHRe). À ces deux mécanismes puissants et relativement lents de contrôle, s’ajoute un système rapide et précis de transfert et d’échange d’ions, d’eau ou de toxines assuré par les cellules gliales. Ces cellules gliales, surtout macrogliales (astrocytes et plus particulièrement les cellules de Müller), sont aussi responsables de la cohésion rétinienne, quand les cellules microgliales en assurent la défense avec une variété de réponses inflammatoires possibles au niveau de la rétine.
La barrière hémato-rétinienne interne commence à être mieux connue, ainsi que ses mécanismes de régulation (voir chapitre 4.2). Sa rupture, ou autrement dit son dysfonctionnement traduit par des transferts anormalement élevés vers la rétine, est la cause la plus reconnue de l’œdème maculaire. Cette rupture peut être due à diverses maladies allant du diabète aux occlusions veineuses. Cette barrière, afin d’être restaurée, est la cible directe de plusieurs médicaments utilisés pour le traitement des œdèmes maculaires indépendamment de la maladie causale.
Un dysfonctionnement de la barrière hémato-rétinienne externe, moins connue, peut aussi causer un œdème maculaire. Elle peut se voir dans de nombreuses maladies seule ou associée à la rupture de la barrière hémato-rétinienne interne (voir chapitre 4.3). C’est aussi une partie plutôt ignorée de la physiopathologie des néovaisseaux choroïdiens et de la baisse d’acuité visuelle qui les accompagne. Pourtant c’est sa restauration qui semble être la principale explication de l’amélioration de l’acuité visuelle retrouvée avec les traitements d’aujourd’hui.
La rupture de ces barrières devient surtout un problème lorsque les autres mécanismes de régulation fine de l’homéostasie rétinienne sont débordés. Les cellules gliales sont les principaux acteurs de contrôle et de maintien de cette homéostasie (voir chapitre 4.4). Une fois épuisées, ces cellules dysfonctionnent à leur tour ; elles ne peuvent plus assurer les conditions du milieu intercellulaire adéquates au fonctionnement neuronal ou à la détoxification indispensable à la survie cellulaire.
Cette perte du contrôle du milieu intercellulaire et ses conséquences sur les cellules rétiniennes vont alors provoquer les symptômes fonctionnels de l’œdème maculaire, mais aussi contribuer à des altérations définitives du tissu rétinien (voir chapitre 4.5). Si les cellules ne peuvent plus fonctionner, l’acuité va baisser d’avantage, et si ces cellules continuent à être dans un milieu hostile, elles peuvent disparaître et induire des altérations supplémentaires, compromettant ainsi les chances d’une restauration complète de la vision, même après une guérison éventuelle de la maladie ou le contrôle de la cause.
Au moins deux autres éléments doivent être ajoutés à cette séquence d’événements. Le premier concerne les cytokines, en particulier le vascular endothelial growth factor (VEGF) (voir chapitre 4.6), et les cytokines inflammatoires (voir chapitre 4.7). Ces cytokines, en particulier le VEGF, peuvent avoir un rôle en amont de la rupture de la barrière hémato-rétinienne, mais aussi en aval une fois l’œdème installé, sous forme de réaction inflammatoire à la souffrance rétinienne. Cette réaction à laquelle s’associe aussi une réaction cellulaire, en particulier avec les cellules microgliales, peut ainsi instaurer un cercle vicieux entretenant l’œdème et ses réactions néfastes. Ces cytokines sont aussi la cible des traitements actuels, de manière spécifique avec les anti-VEGF ou moins spécifique avec les corticoïdes.
Le second concerne tous les éléments aggravants qui peuvent dans certaines circonstances s’associer aux éléments causals pour aggraver un œdème maculaire ou même en être l’élément déclenchant. Le plus connu de ces facteurs est le vitré, dont une attache anormale peut influencer l’œdème maculaire, et qui sert de réservoir tant pour les cytokines causales que les médicaments utilisés pour le traitement de l’œdème (voir chapitre 4.8). D’autres facteurs peuvent aussi intervenir comme, en absence de régulation vasculaire appropriée, la pression artérielle.
La rupture de la barrière avec l’épaississement et la perte de transparence de la rétine, le dysfonctionnement et la mort cellulaire progressive vont tous aboutir à une altération des fonctions visuelles. L’altération de la vision est donc multifactorielle dans les œdèmes maculaires. Elle est aussi multiforme et la mesure de l’acuité visuelle peut être enrichie par d’autres mesures permettant de mieux comprendre le handicap causé par l’œdème maculaire (voir chapitre 4.9).
La complexité des mécanismes détaillés dans ce chapitre permet de comprendre facilement pourquoi l’œdème maculaire, malgré sa définition, ne peut être réduit à un simple épaississement de la rétine et à une baisse d’acuité visuelle consécutive. Ainsi les décalages constatés entre la fonction visuelle et l’étude grossière de l’anatomie rétinienne ne sont guère surprenants. Une étude plus fine de l’œdème et de ses conséquences, ainsi qu’une mesure plus précise de la vision pourront peut-être permettre de réduire ce décalage. Ces connaissances ouvrent des perspectives pour une meilleure utilisation des thérapeutiques existantes mais aussi vers la possibilité de nouvelles cibles.
Aujourd’hui, nous pouvons enfin tracer les traits grossiers de la physiopathologie de l’œdème maculaire : une maladie cause une dérégulation de la barrière hémato-rétinienne interne et/ou externe. Cette dérégulation va à son tour submerger les mécanismes de régulation propre de la rétine causant un dysfonctionnement (altération visuelle réversible), puis une mort cellulaire et des dommages tissulaires (altération visuelle irréversible). Certaines réactions à ces dommages et d’autres facteurs annexes vont aussi agir comme facteurs aggravants, ou créer un cercle vicieux entretenant l’œdème. Plus en amont et plus tôt cette chaîne physiopathologique est rompue, plus on peut espérer restaurer la rétine et la fonction visuelle.
DA. Antonetti1
➤ Les barrières hémato-rétiniennes interne (BHRi) et externe (BHRe) assurent le maintien de l’absence de fluide intrarétinien contribuant ainsi à la conservation d’une bonne vision.
➤ La BHRi est constituée des jonctions serrées entre les cellules endothéliales des capillaires rétiniens non fenêtrés, des péricytes et des pieds des cellules macrogliales de la rétine (astrocytes et cellules gliales de Müller). Ces différents composants assurent l’étanchéité de la BHR.
➤ La rupture de la BHRi peut survenir lorsque ces mécanismes de maintien de l’étanchéité sont dépassés. Le VEGF-A est particulièrement impliqué dans l’augmentation de la perméabilité vasculaire. La restauration de cette BHR passe par la récupération de son étanchéité soit par diminution du taux de VEGF-A, soit par renforcement des jonctions serrées entre les cellules endothéliales sous l’action des corticoïdes.
La formation et le maintien BHR sont indispensables au bon fonctionnement visuel. La perte de ces barrières contribue à un grand nombre de pathologies rétiniennes. Il existe une BHRi (que nous détaillerons ici) et une BHRe formée par les cellules de l’épithélium pigmentaire (EP) unies entre elles par des jonctions serrées. Le rôle de ces BHR est d’assurer le maintien d’un environnement spécialisé au sein de la neurorétine. Par ailleurs, de multiples types cellulaires (neurones et cellules gliales) sont requis pour assurer la fonction visuelle ; ils sont organisés au sein de la rétine en différentes couches cellulaires.
La vascularisation rétinienne, comme d’autres systèmes vasculaires au sein du système nerveux central nécessite une barrière hémato-neuronale.
La BHRi contrôle les échanges de métabolites et de déchets entre la lumière des capillaires et vaisseaux rétiniens, et la neurorétine. Elle est formée par l’interaction de cellules gliales rétiniennes (pied des cellules gliales de Müller et des astrocytes), les péricytes et les cellules endothéliales (fig. 4-1). La BHRi contrôle la perméabilité entre le secteur plasmatique et le secteur neurorétinien et consiste en un réseau jonctionnel complexe entre les cellules endothéliales (capillaires rétiniens non fenestrés). La BHRi permet de maintenir un environnement rétinien approprié au fonctionnement neuronal correct. Les péricytes renforcent les cellules endothéliales par la sécrétion d’angiopoïétine 1 [1], qui favorise l’expression des protéines constituant les jonctions serrées.
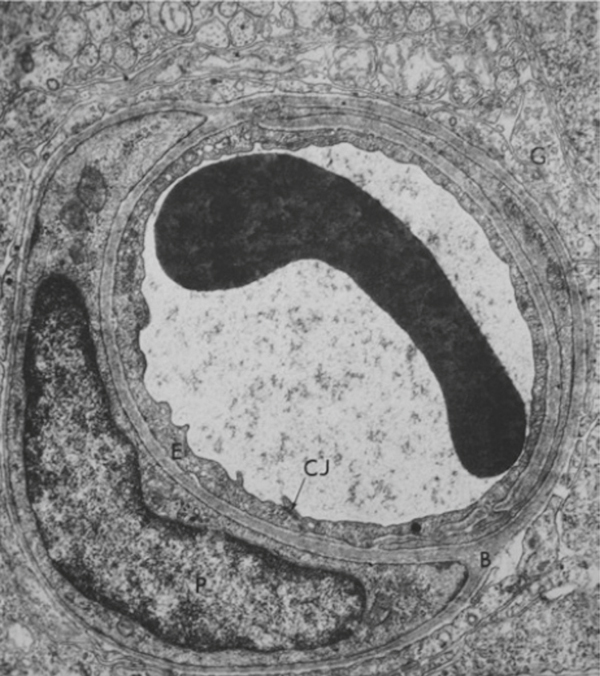
Fig. 4-1 Coupes de capillaire rétinien de rat vu en microscopie électronique représentant les différents composants de la BHRi avec une hématie visible à l’intérieur du capillaire rétinien.
Membrane basale (B), cellule endothéliale (E), péricyte intramural (P) et cellules gliales (G) composées des extensions des cellules gliales de Müller et des astrocytes rétiniens et jonctions cellulaires (CJ) [42].
(Source : Runkle EA, Antonetti DA. The Blood-Brain and Other Neural Barriers. Methods in Molecular Biology 2011 ; 686 : 133-48. © Springer Science+Business Media, LLC 2011. Reproduction autorisée.)
La BHRi est composée de complexes de jonctions serrées et adhérentes (fig. 4-2). Les cellules endothéliales rétiniennes possèdent des systèmes de jonctions serrées bien développés qui leur confèrent un haut degré de contrôle de la perméabilité des fluides et des solutés. Les jonctions serrées restreignent le flux de nombreuses substances telles que les lipides et les protéines [2, 3]. Les capillaires rétiniens sont relativement imperméables, même aux particules aussi petites que les ions sodium [4]. Les jonctions adhérentes sont essentielles au développement de la barrière et influencent la formation des jonctions serrées [5–8]. Ensemble les jonctions serrées et adhérentes créent une barrière protectrice du parenchyme neuronal.
Dans la rétine humaine, l’expression des jonctions serrées débute à la 24e semaine de grossesse [9]. Au cours du temps, l’expression des protéines des jonctions serrées augmente dans le cerveau, dans les vaisseaux rétiniens et dans les cellules de l’EP, aboutissant à une diminution de la perméabilité et l’établissement des barrières hémato-cérébrales et hémato-rétiniennes [10–16]. Les jonctions serrées comportent deux fonctions : celle de filtre qui contrôle le passage de fluide, et celle de barrière qui empêche les mouvements des protéines et des lipides entre le pôle apical et basolatéral des cellules endothéliales. Les jonctions serrées sont composées de plus de 40 protéines, à la fois transmembranaires et intracellulaires. Les protéines transmembranaires comprennent les occludines, la tricelluline, la famille des claudines et les molécules d’adhésion jonctionnelle (junctional adhesion molecules ou JAM). Les protéines transmembranaires sont reliées au cytosquelette par la famille des zonulae occludens (ZO).
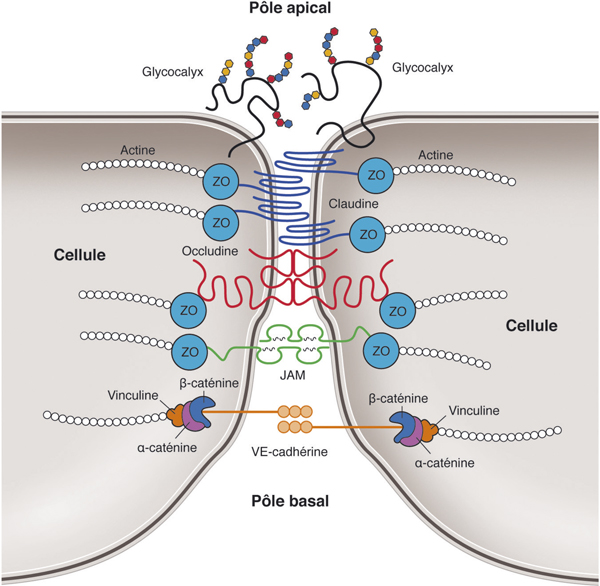
Fig. 4-2 Modèle de BHRi composée de jonctions serrées et de jonctions adhérentes.
Le haut de la figure représente la lumière du capillaire rétinien. Les jonctions serrées correspondent aux protéines transmembranaires claudines, occludines et JAM associées aux protéines architecturales ZO, elles-mêmes connectées à l’actine (au pôle apical des cellules). À l’opposé, les jonctions adhérentes sont concentrées au pôle basal des cellules, et regroupent les VE-cadhérines (cadhérines de l’endothélium vasculaire), associées à la β-caténine et à l’α-caténine qui connecte la vinculine puis l’actine.
(D’après Runkle EA, et al. [42].)
La famille des claudines comprend 24 types protéiques de poids moléculaire moyen compris entre 20 et 27 KDa, avec quatre domaines transmembranaires et deux boucles extracellulaires, avec des extrémités N et C terminales intracellulaires.
Les claudines 1, 5 et 15 sont présentes au niveau des cellules endothéliales et contribuent à la BHRi [17, 18]. Dans des conditions d’hypoxie, l’expression rétinienne de la claudine 5 est réduite de 59 % et la perméabilité vasculaire est augmentée [19].
L’occludine a été la première protéine identifiée des jonctions serrées. Cette protéine est composée d’une terminaison N-terminale intracytoplasmique, quatre domaines transmembranaires, deux boucles extracellulaires, une boucle intracellulaire et une extremité C-terminale. L’occludine est une protéine de poids moléculaire d’environ 59 KDa qui contribue au contrôle des propriétés de barrière étanche et au contrôle du trafic des protéines de jonction dans les cellules endothéliales rétiniennes.
L’analyse spectrométrique a permis de mettre en évidence plusieurs sites de phosphorylation au niveau des occludines dans les cellules endothéliales rétiniennes bovines (BREC) traitées par du VEGF-A [20]. Cette phosphorylation conduit à une ubiquitination (fixation de molécules d’ubiquitine, qui sont un marqueur des protéines à éliminer) des occludines et une endocytose des jonctions serrées.
Une mutation de l’un de ces sites de phosphorylation a permis d’empêcher l’endocytose des protéines de jonctions serrées induite par le VEGF, et a bloqué l’augmentation induite par le VEGF de la perméabilité vasculaire [21]. Ceci confirme le lien étroit entre la présence de VEGF et la perturbation de ces systèmes de jonctions étanches.
Les molécules d’adhésion jonctionnelle (junctional adhesion molecules ou JAM) comprennent un seul domaine transmembranaire. L’extrémité C-terminale est intracellulaire et l’extrémité N-terminale extracellulaire contient deux domaines « immunoglobuline-like » (mimant une immunoglobuline) [22]. Les JAM, composées de JAM-A, -B et -C, interagissent directement avec ZO-1 et partitioning defective 3 (Par-3). Par-3 forme le complexe de polarité en interagissant avec la protéine kinase C atypique (atypical protein kinase C ou aPKC) et se fixant à Par-6. Cette polarité est nécessaire pour la formation et le maintien de ces JAM dans les cellules épithéliales.
Les zonulae occludens (ZO) connectent les protéines transmembranaires des jonctions serrées au cytosquelette. En se liant à d’autres protéines ZO, elles contribuent à la création d’un réseau de jonctions. ZO-1 (210–225 KDa) a été la première protéine de jonction serrée identifiée suivie de ZO-2 (180 KDa) et ZO-3 (130 KDa) [23–27]. Dans les cellules dépourvues de jonctions serrées, ZO-1 et ZO-2 s’associent avec les jonctions adhérentes [28].
Les protéines ZO sont essentielles pour l’organisation des jonctions serrées. L’absence de calcium induit un désassemblage de ce complexe jonctionnel et permet un réassemblage lorsqu’il est réintroduit. L’utilisation d’un système cellulaire qui n’a pas de ZO-1, ZO-2 et ZO-3 révèle que la famille ZO est essentielle pour la formation de la BHR en dirigeant l’insertion de claudines dans la membrane [29]. In vivo, la délétion des gènes codant pour les ZO-1 [30] et ZO-2 [31] est létale au début de l’embryogenèse de la souris. Cependant, des phénotypes distincts suggèrent des fonctions non redondantes pour ces isoformes. La délétion du gène codant pour ZO-1 entraîne des défauts de développement embryonnaire chez la souris, du sac vitellin et de la membrane vasculaire embryonnaire, suggérant son rôle dans l’angiogenèse [30].
L’élévation du VEGF altère l’intégrité de la BHRi dans de nombreuses conditions pathologiques. Une grande partie de ce qui est connu sur ce phénomène se rapporte à la rupture du complexe de jonctions serrées. L’augmentation de la perméabilité paracellulaire est associée à une réduction ou à une redistribution des occludines [32]. Les modèles animaux de diabète démontrent une diminution du nombre d’occludines rétiniennes et une perméabilité vasculaire accrue [33]. Le VEGF-A a pour effet d’affaiblir les jonctions serrées des cellules endothéliales, et augmente également la perméabilité transcellulaire par le biais des cavéoles (vésicules qui proviennent d’une invagination de la membrane plasmique). Elles sont constituées d’un radeau lipidique et d’une protéine transmembranaire, la cavéoline [34]. Hofman et al. [33] ont examiné la perméabilité vasculaire rétinienne après injection intravitréenne de VEGF-A chez le singe, à l’aide d’angiographies à la fluorescéine. Ils ont constaté que le VEGF-A induisait une augmentation de diffusion de colorant ainsi que du nombre de cavéoles.
Astrocytes, cellules gliales de Müller et péricytes coopèrent afin d’induire les différents composants de la BHR permettant le maintien de son étanchéité. L’expression de la protéine kinase A d’ancrage 12 (AKAP12) dans les astrocytes améliore la formation de la BHR par augmentation de l’angiopoïétine 1 (Ang1) et la diminution de VEGF-A [9]. Le facteur induit par l’hypoxie, HIF-1α, est un important facteur de transcription associé à une augmentation de l’expression du VEGF-A liée à l’hypoxie [35]. Ang1 est un ligand pour le récepteur Tie2 et l’association Ang1 avec Tie2 stabilise les vaisseaux sanguins et protège contre la néovascularisation et la perméabilité induite par une production excessive de VEGF-A [36–38]. Ces données suggèrent un signal moléculaire provenant des astrocytes, contrôlé en partie par AKAP12, qui induit l’expression des jonctions serrées dans l’endothélium vasculaire et est nécessaire pour le maintien d’une barrière fonctionnelle.
Les glucocorticoïdes ont également un effet important sur la barrière endothéliale. Le récepteur des glucocorticoïdes (glucocorticoid receptor ou GR) est maintenu dans un état inactif dans le cytoplasme. L’activation du GR favorise la fonction de barrière en augmentant la teneur en protéines des jonctions serrées [39] et la redistribution de ces protéines vers la membrane cytoplasmique des cellules endothéliales [40]. Les propriétés de barrière induites par les glucocorticoïdes ont été confirmées in vivo [41]. Fait intéressant, les glucocorticoïdes inversent la phosphorylation des occludines et favorisent ainsi la restauration de la perméabilité vasculaire [39].
La rétine est un tissu neuronal hautement spécialisé nécessitant une structure vasculaire unique et un contrôle strict de la perméabilité pour permettre une bonne fonction visuelle. La réglementation des flux de diffusion hématogène des métabolites dans la rétine est contrôlée par l’EP (BHRe) et par les vaisseaux sanguins rétiniens spécialisés, qui utilisent un système complexe de jonctions (BHRi) pour réguler la perméabilité et maintenir l’environnement des neurones de la rétine.
Notre compréhension actuelle de la BHRi implique les claudines dans la formation de la barrière, les occludines dans la régulation de la perméabilité, et les ZO dans l’architecture de la barrière par interaction avec des protéines transmembranaires. Cependant, une foule de protéines de jonction supplémentaires ont été identifiées et leur rôle dans la qualité de la BHR reste encore mal défini. En outre, la fonction des cellules gliales (voir chapitre 4.7) et des péricytes pour induire et maintenir la BHRi a été clairement démontrée, mais nous commençons tout juste à comprendre l’interaction complexe entre ces multiples types cellulaires. Les études futures élucidant l’induction et le maintien de la BHR apporteront une meilleure compréhension dans le fonctionnement des différentes molécules utilisées pour restaurer la BHR lorsqu’elle est compromise au cours des maladies rétiniennes.
[1] Hori S, Ohtsuki S, Hosoya K, et al. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem 2004 ; 89 : 503-13.
[2] Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol 1963 ; 17 : 375-412.
[3] Van Meer G, Simons K. The function of tight junctions in maintaining differences in lipid composition between the apical and the basolateral cell surface domains of MDCK cells. EMBO J 1986 ; 5 : 1455-64.
[4] Törnquist P, Alm A, Bill A. Permeability of ocular vessels and transport across the blood-retinal-barrier. Eye 1990 ; 4 (Pt 2) : 303-9.
[5] Fukuhara A, Irie K, Nakanishi H, et al. Involvement of nectin in the localization of junctional adhesion molecule at tight junctions. Oncogene 2002 ; 21 : 7642-55.
[6] Fukuhara A, Irie K, Yamada A, et al. Role of nectin in organization of tight junctions in epithelial cells. Genes Cells Devoted Mol Cell Mech 2002 ; 7 : 1059-72.
[7] Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev 2005 ; 57 : 815-55.
[8] Suzuki A, Ishiyama C, Hashiba K, et al. aPKC kinase activity is required for the asymmetric differentiation of the premature junctional complex during epithelial cell polarization. J Cell Sci 2002 ; 115 : 3565-73.
[9] Choi YK, Kim JH, Kim WJ, et al. AKAP12 regulates human blood-retinal barrier formation by downregulation of hypoxia-inducible factor-1alpha. J Neurosci Off J Soc Neurosci 2007 ; 27 : 4472-81.
[10] Dermietzel R, Krause D. Molecular anatomy of the blood-brain barrier as defined by immunocytochemistry. Int Rev Cytol 1991 ; 127 : 57-109.
[11] Hirase T, Staddon JM, Saitou M, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci 1997 ; 110 (Pt 14) : 1603-13.
[12] Rizzolo LJ. Polarity and the development of the outer blood-retinal barrier. Histol Histopathol 1997 ; 12 : 1057-67.
[13] Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci 1999 ; 22 : 11-28.
[14] Stewart PA, Hayakawa K. Early ultrastructural changes in blood-brain barrier vessels of the rat embryo. Brain Res Dev 1994 ; 78 : 25-34.
[15] Williams CD, Rizzolo LJ. Remodeling of junctional complexes during the development of the outer blood-retinal barrier. Anat Rec 1997 ; 249 : 380-8.
[16] Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier : development, composition and regulation. Vascul Pharmacol 2002 ; 38 : 323-37.
[17] Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci 1999 ; 96 : 511-6.
[18] Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin : claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 1999 ; 147 : 185-94.
[19] Koto T, Takubo K, Ishida S, et al. Hypoxia disrupts the barrier function of neural blood vessels through changes in the expression of claudin-5 in endothelial cells. Am J Pathol 2007 ; 170 : 1389-97.
[20] Sundstrom JM, Tash BR, Murakami T, et al. Identification and analysis of occludin phosphosites : a combined mass spectrometry and bioinformatics approach. J Proteome Res 2009 ; 8 : 808-17.
[21] Phillips BE, Cancel L, Tarbell JM, Antonetti DA. Occludin independently regulates permeability under hydrostatic pressure and cell division in retinal pigment epithelial cells. Invest. Ophthalmol Vis Sci 2008 ; 49 : 2568-76.
[22] Chiba H, Osanai M, Murata M, et al. Transmembrane proteins of tight junctions. Biochim Biophys Acta 2008 ; 1778 : 588-600.
[23] Balda MS, Gonzalez-Mariscal L, Matter K, et al. Assembly of the tight junction : the role of diacylglycerol. J Cell Biol 1993 ; 123 : 293-302.
[24] Gumbiner B, Lowenkopf T, Apatira D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc Natl Acad Sci 1991 ; 88 : 3460-4.
[25] Itoh M, Morita K, Tsukita S. Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J Biol Chem 1999 ; 274 : 5981-6.
[26] Jesaitis LA, Goodenough DA. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J Cell Biol 1994 ; 124 : 949-61.
[27] Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1 : a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol 1986 ; 103 : 755-66.
[28] Willott E, Balda MS, Heintzelman M, et al. Localization and differential expression of two isoforms of the tight junction protein ZO-1. Am J Physiol 1992 ; 262 : C1119-24.
[29] Katsuno T, Umeda K, Matsui T, et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell 2008 ; 19 : 2465-75.
[30] Xu J, Kausalya PJ, Phua DCY, et al. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol 2008 ; 28 : 1669-78.
[31] Antonetti DA, Barber AJ, Hollinger LA, et al. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem 1999 ; 274 : 23463-7.
[32] Antonetti DA, Barber AJ, Khin S, et al. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content : vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 1998 ; 47 : 1953-9.
[33] Hofman P, Blaauwgeers HG, Tolentino MJ, et al. VEGF-A induced hyperpermeability of blood-retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Vascular endothelial growth factor-A. Curr Eye Res 2000 ; 21 : 637-45.
[34] Feng Y, Venema VJ, Venema RC, et al. VEGF-induced permeability increase is mediated by caveolae. Invest Ophthalmol Vis Sci 1999 ; 40 : 157-67.
[35] Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997 ; 277 : 55-60.
[36] Asahara T, Chen D, Takahashi T, et al. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res 1998 ; 83 : 233-40.
[37] Romero IA, Radewicz K, Jubin E, et al. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neurosci Lett 2003 ; 344 : 112-6.
[38] Thurston G, Rudge JS, Ioffe E, et al. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med 2000 ; 6 : 460-3.
[39] Antonetti DA, Wolpert EB, Demaio L, et al. Hydrocortisone decreases retinal endothelial cell water and solute flux coincident with increased content and decreased phosphorylation of occludin. J Neurochem 2002 ; 80 : 667-77.
[40] Edelman JL, Lutz D, Castro MR. Corticosteroids inhibit VEGF-induced vascular leakage in a rabbit model of blood-retinal and blood-aqueous barrier breakdown. Exp Eye Res 2005 ; 80 : 249-58.
[41] Felinski EA, Cox AS, Phillips BE, Antonetti DA. Glucocorticoids induce transactivation of tight junction genes occludin and claudin-5 in retinal endothelial cells via a novel cis-element. Exp Eye Res 2008 ; 86 : 867-78.
[42] Runkle EA, Antonetti DA. The blood-brain and other neural barriers. Methods in Molecular Biology 2011 ; 686 : 133-48.
F. Behar-Cohen, A. Daruich, A. Matet, P. Crisanti-Lassiaz
➤ La barrière hémato-rétinienne externe (BHRe) est formée par des zonulae occludens (jonctions serrées, sans espace intercellulaire détectable) et des zonulae adherens (jonctions adhérentes, maintenant un espace intercellulaire) situées entre les cellules de l’épithélium pigmentaire de la rétine.
➤ La membrane limitante externe participerait aussi à la fonction de barrière.
➤ La BHRe a pour rôle principal de contrôler les échanges hydro-ioniques et métaboliques entre la choriocapillaire et la rétine externe, tout en garantissant le privilège immunitaire de l’œil et en limitant le passage de substances toxiques.
➤ La perte de fonction de barrière peut résulter de différents mécanismes plus ou moins intriqués : dysfonction des cellules de l’épithélium pigmentaire, déstabilisation des jonctions serrées, altérations de la membrane limitante externe.
➤ Les mécanismes impliqués diffèrent en fonction des étiologies de l’œdème maculaire.
L’œdème maculaire (OM) peut être défini comme un excès de fluide dans la neurorétine et/ou dans l’espace sous-rétinien, au niveau de la macula. Il peut se manifester par un épaississement diffus de la rétine, la formation de kystes intrarétiniens et l’accumulation de liquide sous-rétinien. La localisation de l’accumulation de fluide ne permet pas d’en déduire les mécanismes physiopathogéniques. L’OM peut survenir dans la quasi-totalité des pathologies rétiniennes, à diverses phases de leur évolution, et résulte fréquemment de processus métaboliques, ischémiques/hypoxiques et inflammatoires.
La barrière hémato-rétinienne externe (BHRe) est formée principalement par l’épithélium pigmentaire de la rétine (EPR), couche monocellulaire à jonctions intercellulaires serrées [1]. La BHRe a pour rôle principal de contrôler les échanges hydro-ioniques et métaboliques entre la choriocapillaire et la rétine externe, tout en garantissant le privilège immunitaire de l’œil et en limitant le passage de substances toxiques. Dans une moindre mesure, la membrane limitante externe (MLE) participe également à la fonction de barrière externe. Celle-ci est constituée par des jonctions cellulaires entre cellules de Müller et photorécepteurs ; elle limite la diffusion de fluide entre l’espace sous-rétinien et la rétine interne. Enfin, les jonctions cellulaires serrées des cellules endothéliales de la choriocapillaire et ses fenestrations diaphragmées contribuent également aux transports sélectifs et contrôlés (fig. 4-3).
En conditions physiologiques, les flux entrant dans la rétine proviennent du vitré, de la circulation rétinienne (contrôlée par la BHRi, formée par les jonctions serrées entre les cellules endothéliales des capillaires rétiniens) et de la choriocapillaire, cette dernière voie étant contrôlée par la BHRe et impliquant des flux traversant l’espace sous-rétinien. De même, une des voies principales de drainage du liquide rétinien repose sur sa résorption par l’EPR et son élimination vers la choroïde. Ces flux à travers la BHRe reposent sur des canaux hydro-ioniques actifs et strictement régulés, qui contribuent à l’homéostasie et la transparence optique des milieux rétiniens (fig. 4-4) [2]. Leur fonctionnement sera abordé dans ce sous-chapitre, ainsi que les mécanismes de dérégulation par des facteurs locaux ou systémiques conduisant à un OM.

Fig. 4-3 Complexe épithélium pigmentaire–choroïde.
a. Coupe histologique de la rétine humaine montrant les relations entre les cellules de l’EPR et les photorécepteurs sus-jacents et la choriocapillaire sous-jacente. b. Microscopie électronique à transmission montrant des fenestrations dans les cellules endothéliales de la choriocapillaire et les diaphragmes présents dans ces fenestrations qui mesurent environ 700–800 A. c. Microscopie électronique à balayage d’un moulage de la vascularisation choroïdienne montrant la choriocapillaire et les gros vaisseaux choroïdiens. d. Microscopie électronique à transmission montrant la fusion de la membrane basale de la choriocapillaire avec la membrane de Bruch. e. Microscopie électronique à transmission de la face basale et des villosités de l’EPR, de la membrane de Bruch et des fenestrations des cellules endothéliales de la choriocapillaire dont on distingue un diaphragme (flèche orange).
CC : choriocapillaire ; CNE : couche nucléaire externe ; EPR : épithélium pigmentaire de la rétine ; GVC : gros vaisseaux choroïdiens ; MB : membrane de Bruch ; MLE : membrane limitante externe ; SE : segments externes des photorécepteurs ; SI : segment interne des photorécepteurs.
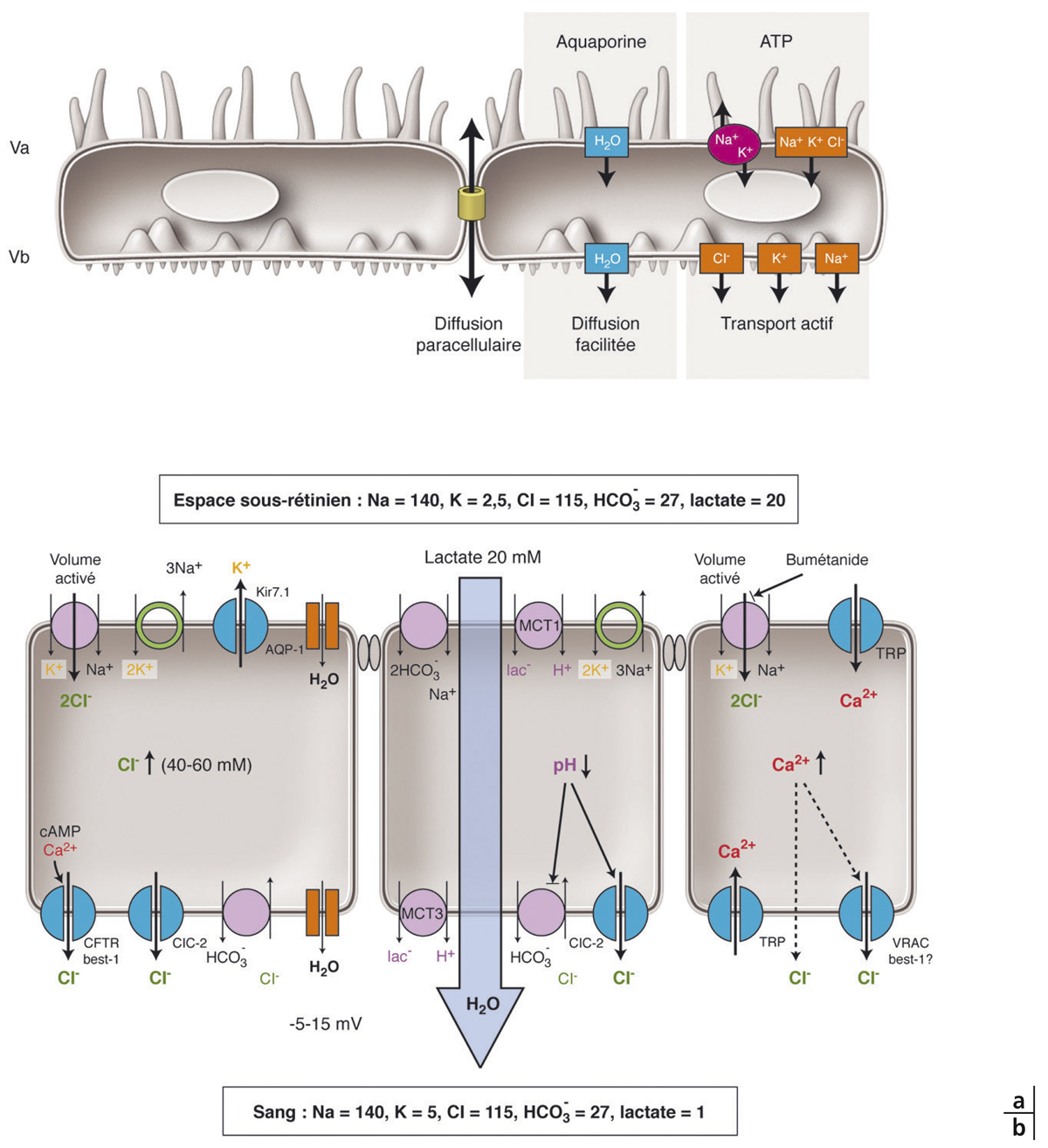
Fig. 4-4 Transport transépithélial.
a. Représentation schématique de l’EPR avec ses villosités apicales (Va) et basales (Vb), son complexe jonctionnel apical (cylindre orange) et les différents mécanismes de transport transcellulaire. b. Mécanismes du transport transépithélial actif. À gauche : le transport d’eau et de chlore s’effectue du pôle apical au pôle basal grâce à un transport actif de Na+/K+/2Cl–. L’eau suit le transport de Cl– par des canaux à eau. Au milieu : les changements de pH (régulés par les transporteurs Na+/HCO3- et Cl–/HCO3-) activent les transporteurs monocarboxylate (MCT1 et MCT3) qui transportent le lactate et l’eau à travers les cellules d’EPR. À droite : des canaux Na+/K+/2Cl– et Cl– sensibles au changement de volume des cellules induisent des modifications de calcium intracellulaire avec une probable induction de canaux TRP (transient receptor potential channel) et de canaux K+ dépendant du calcium.
(D’après Reichhart N, Strauss O. Ion channels and transporters of the retinal pigment epithelium. Exp Eye Res 2014 ; 126 : 27-37.)
L’EPR est une monocouche de 4 à 6 millions de cellules hexagonales [3], qui s’étend du nerf optique jusqu’à l’ora serrata, prolongé en avant par l’épithélium pigmenté du corps ciliaire. Les cellules de l’EPR sont plus denses et plus hautes au niveau de la fovéa. L’EPR occupe une situation stratégique, séparant les photorécepteurs (à sa face interne) de la membrane de Bruch et la choroïde (à sa face externe). La membrane basale de l’épithélium pigmentaire forme la couche la plus interne de la membrane de Bruch. Les cellules de l’EPR possèdent une polarité dans leur composition et dans les fonctions assurées par leurs pôles apical et basal (fig. 4-5). La face apicale présente de nombreuses villosités en contact étroit avec les segments externes des photorécepteurs, permettant d’assurer leur phagocytose et le recyclage d’une partie des photopigments. Une seule cellule de l’EPR se trouve en effet au contact de 35 à 45 segments externes (fig. 4-5). Ces interdigitations, en conjonction avec la matrice extracellulaire et des molécules d’adhésion telles que N-CAM (neural cell adhesion molecule) participent à l’adhésion relative entre l’EPR et la rétine neurosensorielle, séparés par un espace virtuel, l’espace sous-rétinien. Plus de 280 protéines participant à la phagocytose des segments externes des photorécepteurs ont été identifiées au sein de ces microvillosités dont des enzymes du cytosquelette, des composants de la matrice extracellulaire, des transporteurs et des intégrines [4–6]. Le canal Na/K/ATPase, principalement exprimé à la face apicale des cellules de l’EPR, assure le transport ionique transépithélial, critique pour la fonction de pompe de l’EPR (voir fig. 4-4). Une autre protéine de la membrane apicale, chloride intracellular channel 4 (CLIC4), est impliquée dans la régulation du transport ionique et l’adhésion entre EPR et photorécepteurs.
À la membrane basale de l’EPR se trouvent des intégrines (α3β1, α6β1 et αvβ3) qui participent à l’attachement des cellules à la membrane de Bruch, ainsi que la bestrophine-1 (Best-1), un canal au chlore calcium-dépendant. Certains transporteurs se trouvent aux deux faces des cellules de l’EPR, comme le transporteur du glucose GLUT1, tandis que certaines fonctions sont assurées par des transporteurs différents à la face apicale et basale de la cellule, telle que l’élimination du lactate depuis l’espace sous-rétinien assurée respectivement par MCT1 (monocarboxylate transporter 1) et MCT3 (voir fig. 4-4) [7].
La distribution intracellulaire des organelles présente également une polarité en rapport avec les fonctions de l’EPR. Dans la partie apicale de la cellule, à proximité des microvillosités responsables de la phagocytose des segments externes, on note la présence de lysosomes contenant des enzymes hydrolytiques, de phagosomes contenant des inclusions lamellaires de membranes de photorécepteurs, et du réticulum endoplasmique. On y trouve aussi des grains de mélanine dont on présume qu’ils absorbent la lumière visible et les ultraviolets, permettant de limiter les dommages rétiniens liés aux radicaux libres, optimisant ainsi la qualité optique en réduisant la dispersion lumineuse. Dans les portions cellulaires centrale et basale se concentrent des grains de lipofuscine, produits par la dégradation des segments externes, ainsi que de nombreuses mitochondries, indiquant la forte dépense énergétique due à la fonction pompe des cellules de l’EPR. Le noyau cellulaire se situe quant à lui dans la partie basale de la cellule (voir fig. 4-5).
La face latérale de la membrane plasmique est pourvue de jonctions intercellulaires, à l’extrémité apicale des cellules. Cette barrière terminale constitue le principal élément de la BHRe (voir fig. 4-5).
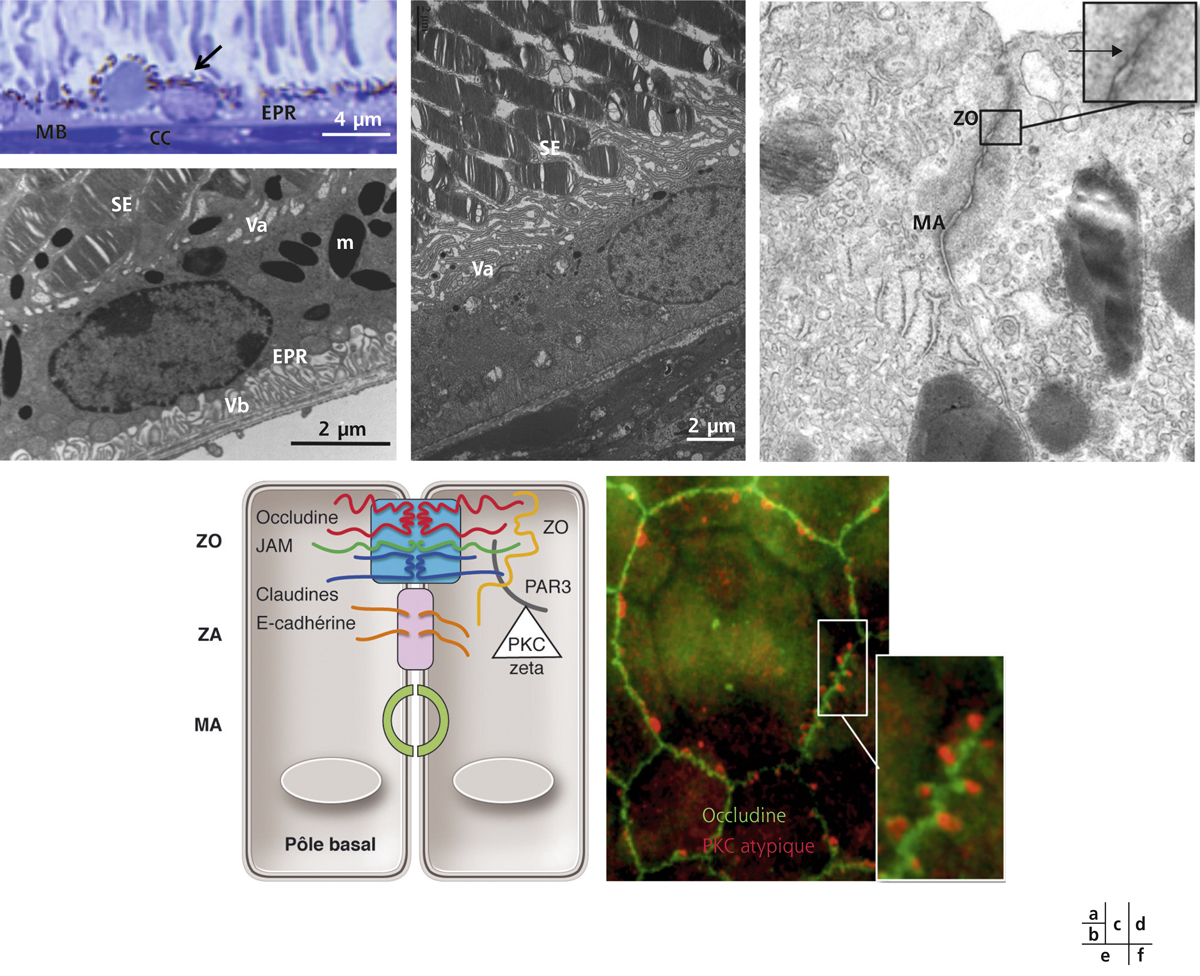
Fig. 4-5 Barrière hémato-rétinienne externe.
a. Coupe histologique de l’épithélium pigmentaire (EPR) montrant les mélanosomes (flèche) en situation apicale et la choriocapillaire (CC) à la face externe de la membrane de Bruch (MB). b. Microscopie électronique à transmission montrant l’EPR de rat avec ses villosités apicales (Va) englobant les segments externes des photorécepteurs (SE) et ses villosités basales (Vb). Les mélanosomes (m) sont en situation apicale. c. Image en microscopie électronique à transmission de l’EPR de rat montrant l’importance des villosités apicales (Va) qui entourent les segments externes des photorécepteurs (SE). d. Image en microscopie électronique à transmission de la jonction intercellulaire montrant la zonula occludens (ZO) (agrandissement en haut à gauche) et la macula adherens (MA). e. Représentation schématique de la jonction apicale épithéliale qui comprend la zonula occludens (ZO), la zonula adherens (ZA) et la macula adherens (MA). f. Image des jonctions intercellulaires de l’EPR monté à plat avec un marquage en vert de l’occludine et en rouge de la PKC-ζ atypique qui se localise dans les boucles de l’occludine en « fermeture éclair ».
La BHRe est principalement formée par des zonulae occludens (jonctions serrées, sans espace intercellulaire détectable) et des zonulae adherens (jonctions adhérentes, maintenant un espace intercellulaire) situées entre les cellules de l’EPR. Ces jonctions appartiennent au groupe des structures membranaires intégrales et sont connectées au cytosquelette d’actine via différentes molécules adaptatrices. Les principaux groupes de protéines qui forment les zonulae occludens sont les claudines (en particulier la claudine-5) et l’occludine. Le domaine cytoplasmique de l’occludine interagit avec des protéines à domaine PDZ, notamment ZO-1 qui régule la prolifération, la différenciation cellulaire et l’homéostasie de l’EPR. Il existe également des JAM (JAM), en particulier JAM-A qui contrôle la polarisation apico-basale et régule la perméabilité de l’épithélium, et JAM-C qui régule le contact intercellulaire via le recrutement de la N-cadhérine et de ZO-1. Il existe aussi des proteine kinases atypiques, indépendantes du calcium, responsables de la phosphorylation des protéines de jonction, comme la protéine kinase C-zeta ou PKC-ζ (voir fig. 4-5).
Les jonctions adhérentes maintiennent une séparation de 20 nm entre les cellules de l’EPR. Les cadhérines sont les principales composantes des jonctions adhérentes (voir fig. 4-5). Ces protéines transmembranaires interagissent avec de nombreuses protéines cytoplasmiques : caténines, α-actine et vinculine. Les jonctions adhérentes initient et maintiennent l’adhésion intercellulaire et la régulation du cytosquelette d’actine, stabilisent la forme polygonale des cellules, et participent à la signalisation intercellulaire et à la régulation transcriptionnelle [8].
Des gap-junctions, laissant un espace intercellulaire de 2 nm, sont aussi présentes dans la membrane latérale des cellules de l’EPR, proches de leur pôle basal. Constituées principalement de connexine, elles jouent un rôle prépondérant dans l’échange d’électrolytes et de métabolites.
Enfin, des desmosomes (macula adherens) ont été occasionnellement décrits entre les cellules de l’EPR chez l’homme, formant des complexes avec les mitochondries. Ils sont potentiellement impliqués dans des fonctions de soutien métabolique et de régulation du calcium intracellulaire [9].
Ces structures jonctionnelles forment une barrière de résistance relativement faible comparée à celle des barrières épithéliales de l’intestin ou de la cornée. Elles nécessitent donc un processus actif et dynamique consommateur d’énergie. La multitude de transporteurs régulés et la présence de protéines d’efflux contribuent au maintien d’une barrière hautement sélective.
Chez les vertébrés, des jonctions adhérentes et des desmosomes ont été observés au niveau de la membrane limitante externe (MLE). En effet, celle-ci n’est pas une « membrane » au sens histologique du terme : il s’agit d’une zone de jonction entre les procès apicaux des cellules de Müller et les segments internes des photorécepteurs. Plus récemment, des protéines JAM, ZO-1 et occludine ont été localisées dans la MLE chez le rat, le singe et l’homme (fig. 4-6), suggérant la présence de jonctions hétérotypiques tight-like dans la MLE [8]. Cette dernière contrôle les mouvements passifs de fluide dans les couches externes de la rétine et possède également une fonction de barrière à la diffusion de protéines [10]. Elle pourrait donc être considérée comme une troisième BHR ou comme partie intégrante de la BHRe.
Au-delà de leur rôle dans l’établissement de la MLE, les cellules gliales de Müller jouent aussi un rôle important dans le drainage d’électrolytes et d’eau depuis la rétine interne vers les vaisseaux rétiniens. En conditions physiologiques, le transport d’ions K+ est en effet associé au drainage des molécules d’eau via les canaux de la famille Kir (K+-inwardly rectifying channels) et les aquaporines (AQP), exprimés par les cellules de Müller [11]. D’autres transporteurs de types glyceroporine jouent un rôle important dans le contrôle des flux hydriques, agissant de façon synchrone pour réguler l’état d’hydratation, l’épaisseur et la transparence de la rétine. En outre, la densité des cellules de Müller est plus élevée dans la macula que dans toute autre région de la rétine. Leur morphologie y diffère également, avec une portion périfovéale orientée de façon radiaire et quasi parallèle au plan frontal, ce qui suggère que les propriétés des cellules de Müller diffèrent entre la macula et la périphérie, et pourrait expliquer la susceptibilité accrue de la région maculaire à l’œdème.
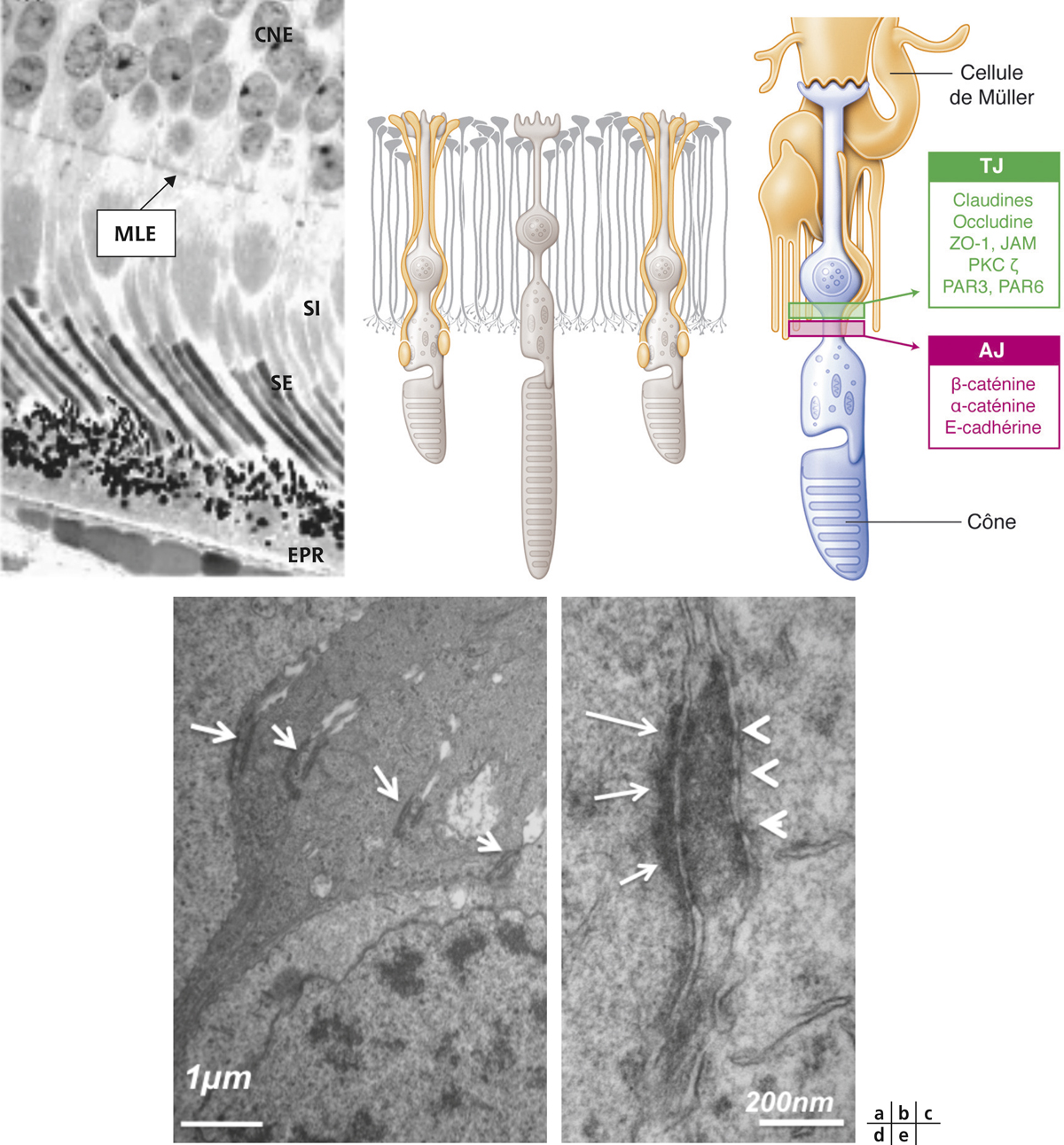
Fig. 4-6 Membrane limitante externe (MLE).
a. Image d’une coupe histologique de rétine humaine montrant la MLE et ses rapports avec la couche nucléaire externe (CNE), les segments internes (SI) et externes (SE) des photorécepteurs et l’EPR. b. Représentation schématique de la MLE montrant les cellules gliales de Müller (en jaune-orange) qui entourent les cônes et forment des jonctions entre leurs apex et la partie proximale du segment interne. c. Représentation schématique des cellules de Müller (en jaune-orange) et d’un cône (en bleu) avec la composition moléculaire des jonctions serrées (TJ) et adhérentes (AJ). d. Image en microscopie électronique à transmission de la MLE montrant les zones de jonctions entre les cellules de Müller et les cônes (flèches blanches). e. Image en microscopie électronique à transmission de la MLE montrant une jonction hétérotypique avec une zone de zonula occludens (tête de flèche) et une zone de macula adherens (flèches).
La dysfonction des cellules de l’EPR elles-mêmes peut contribuer au passage de fluide depuis la choroïde vers l’espace sous-rétinien. Une des fonctions essentielles des cellules de l’EPR est le transport d’eau depuis l’espace sous-rétinien vers la choroïde, sans rupture des jonctions serrées de l’EPR. Il est généralement admis que le transport de Cl– et K+ par les cellules de l’EPR est à l’origine d’un flux hydrique transépithélial. La vitesse de transport de l’eau via les cellules de l’EPR est estimée entre 1,4 et 11 µl/cm2/heure [12]. L’absorption de fluide repose sur des mécanismes complexes se produisant au niveau des membranes apicale et basolatérale des cellules de l’EPR, impliquant le transport de Cl–, l’activité de pompes Na+/K+/ATPase, de canaux ioniques Ca2+-dépendants et volume-dépendants, et de l’adénosine monophosphate (AMP) cyclique. Ces mécanismes, influencés par le rythme circadien, sont régulés de façon différente en conditions photopiques et scotopiques [13–15]. L’absorption d’ions par l’EPR s’accompagne d’un transport hydrique via les aquaporines [16]. Les canaux calciques de l’EPR régulent l’expression de VEGF), suggérant un lien potentiel entre le transport ionique via l’EPR et la perméabilité induite par le VEGF [17].
Dans certaines conditions pathologiques, comme la rétinopathie diabétique, des modifications dans l’expression des aquaporines et des pompes Na+/K+ ont été démontrées au niveau de l’EPR [18]. Dans la choriorétinopathie séreuse centrale, l’accumulation de liquide sous-rétinien résulterait d’une altération des transports de fluide et d’ions à travers l’EPR. Néanmoins, on ne sait pas si ces modifications sont à elles seules capables d’engendrer l’accumulation de liquide sous-rétinien sans dysfonctionnement des jonctions de l’EPR. D’autres pathologies inflammatoires peuvent entraîner des décollements séreux rétiniens du fait d’altérations fonctionnelles des cellules de l’EPR.
Les propriétés de barrière des vaisseaux rétiniens et de l’EPR sont dues principalement à la présence de réseaux complexes de jonctions serrées entre les cellules. La déstabilisation des jonctions serrées peut résulter de multiples facteurs. Elle peut être causée par :
-
une altération de l’activité enzymatique de phosphorylation (par exemple, PKC-ζ au cours du diabète) [19] ;
-
la réduction de l’expression des protéines de jonction (par exemple, l’occludine au cours du diabète ; fig. 4-7) [8] ;
-
des altérations du cytosquelette (par exemple, après dommage oxydatif ou activation de la voie RhoA/ROCK1) ;
-
des modifications des mouvements de calcium [20] ;
-
des dommages ou même des pertes cellulaires (par exemple dans des processus inflammatoires sévères) ;
-
la dégradation moléculaire des jonctions serrées par des protéases activées [21].
En cas d’inflammation, les mécanismes moléculaires précis qui conduisent aux altérations des jonctions serrées restent imparfaitement compris.
Plusieurs voies de signalisation extracellulaires conduisant à une phosphorylation de l’actine et/ou de protéines de jonction pourraient aussi intervenir, provoquant leur déplacement de la membrane cytoplasmique vers d’autres compartiments intracellulaires. Le stress mécanique pourrait aussi contribuer à la rupture des jonctions serrées comme observé lorsque l’EPR est soumis à une hyperpression chronique, que ce soit en cas de tumeur vasculaire ou mélanocytique volumineuse dans la choroïde ou en cas de vasodilatation choroïdienne dans la choriorétinite séreuse centrale. La perméabilité de l’EPR est aussi favorisée par de nombreux médiateurs solubles tels que :
-
des cytokines : monocyte chemoattractant protein-1 (MCP-1) ; tumor-necrosis factor-α (TNF-α) ; interleukine (IL) 1b, 8, 6 ; interféron γ ;
-
des membres de la famille VEGF : VEGF-A, VEGF-B, VEGF-R1 ;
-
des systèmes d’activation plasmatique : facteurs du complément, facteurs de la coagulation, facteurs de fibrinolyse ;
-
des amines vaso-actives : histamine, sérotonine ;
-
des métabolites de l’acide arachidonique : prostaglandine E2 (PGE2) ;
-
des radicaux libres oxygénés.
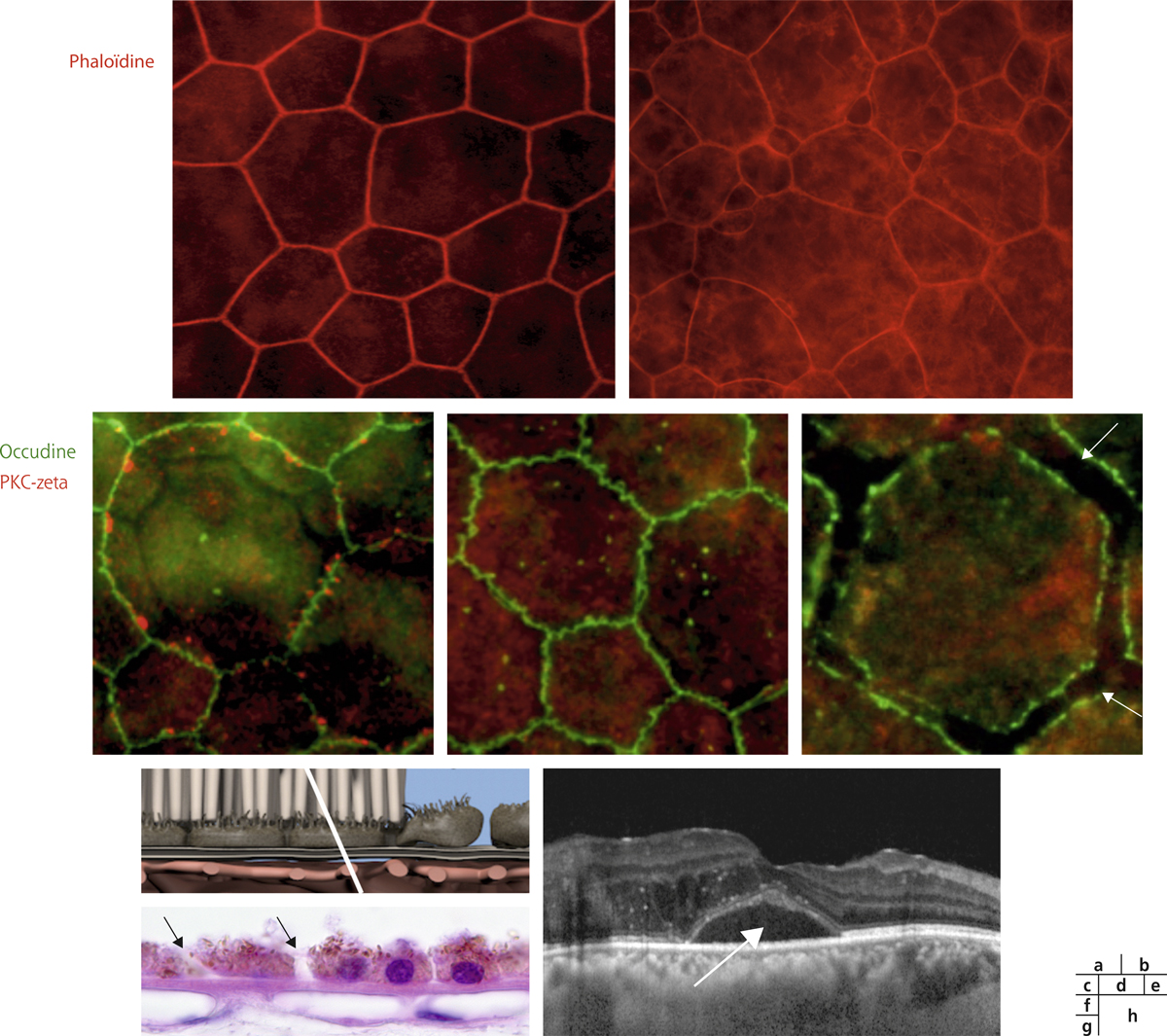
Fig. 4-7 Altérations de la barrière externe au cours du diabète.
a. Montage à plat de l’épithélium pigmentaire de la rétine (EPR) de rat normal avec visualisation des contours cellulaires par la phaloïdine. b. Montage à plat de l’EPR d’un rat diabétique de type 2 après 3 mois de diabète montrant une altération de la structure du cytosquelette. c. Marquage des jonctions serrées de l’EPR d’un rat normal. d. Perte de la localisation de la PKC-ζ dans les boucles de l’occludine après 4 mois de diabète. e. Ouverture des jonctions serrées de l’EPR après 1 an de diabète. f. Représentation schématique d’un EPR normal et d’un EPR avec jonctions rompues. g. Image en microscopie d’un EPR chez un patient diabétique sans rétinopathie cliniquement connue montrant une ouverture des jonctions serrées de l’EPR (flèches). h. Image en SD-OCT d’un patient avec œdème maculaire diabétique et présence de liquide sous-rétinien (flèche) témoignant d’une rupture de la BHRe.
(Source figure 4-7g : remerciements au Dr A. Moulin, Laboratoire de pathologie oculaire, hôpital ophtalmique Jules-Gonin, Lausanne, Suisse.)
Des anomalies de la MLE pourraient jouer un rôle dans la physiopathologie de l’œdème maculaire. Dans un modèle de diabète chez le rat, une dissociation des jonctions de la MLE ainsi qu’une diminution de l’occludine au niveau de la MLE a été observée (fig. 4-8). Celle-ci était associée à un gonflement des cellules de Müller et à la formation de kystes intrarétiniens [8]. En outre, une corrélation positive a été mise en évidence entre l’importance de la rupture de la MLE, objectivée en tomographie à cohérence optique et les taux vitréens de VEGF et de intercellular adhesion molecule 1 (ICAM-1) chez les patients diabétiques, ces deux cytokines étant fortement associées à l’œdème maculaire [22].
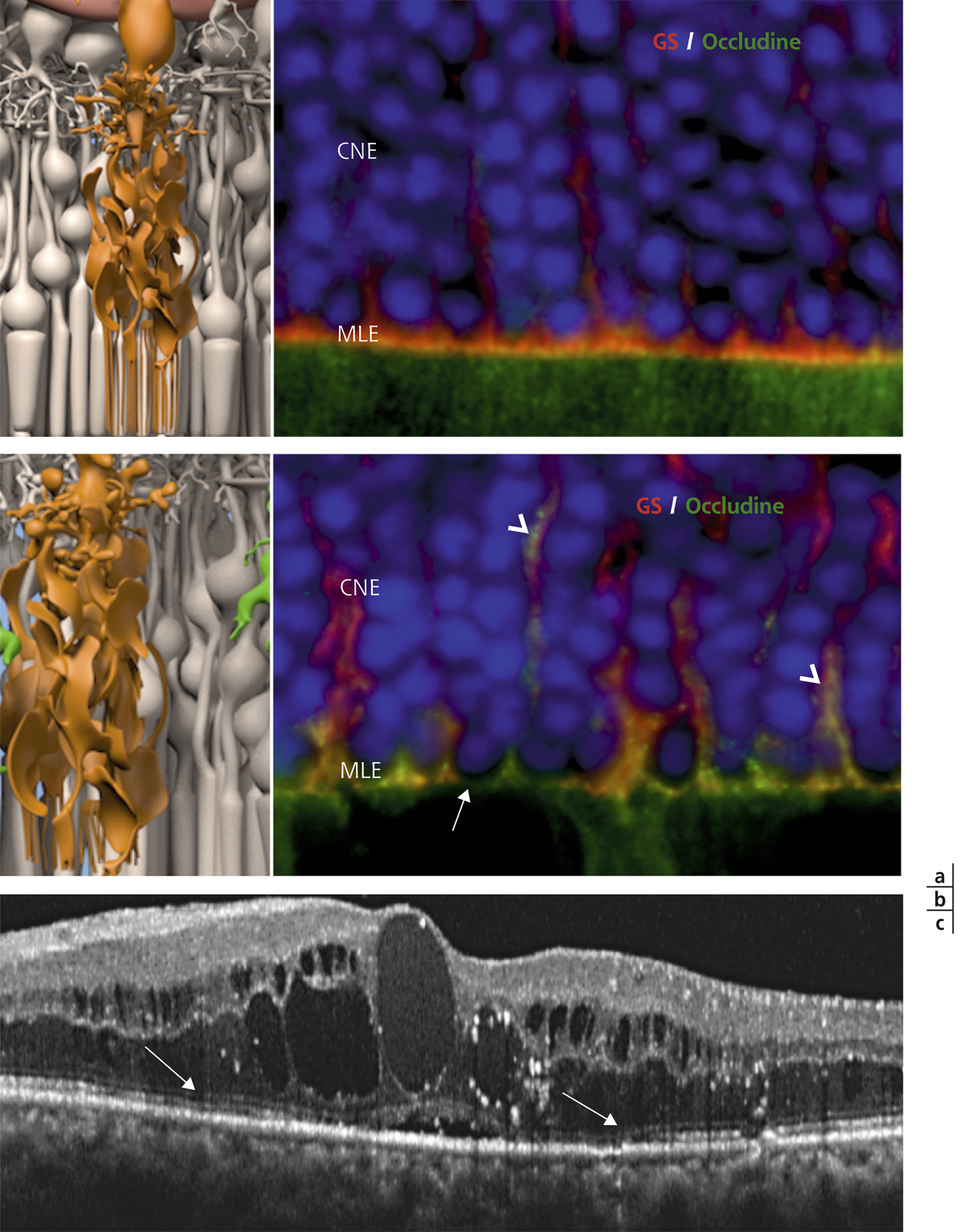
Fig. 4-8 Modifications de la MLE au cours du diabète.
a. MLE en conditions normales montrant un co-marquage entre les apex des cellules de Müller (marquées avec la glutamine synthétase ou GS) et l’occludine. b. Chez un rat diabétique de 1 an, on observe des ruptures de la MLE avec perte de la localisation de l’occludine (flèche blanche) qui se déplace dans les corps cellulaires des Müller œdématiées (tête de flèche). c. Œdème maculaire visualisé en SD-OCT chez un patient diabétique montrant que la MLE est discontinue par endroits (flèches).
-
Les mécanismes conduisant à un OM sont souvent multiples et impliquent de façon intriquée l’altération des BHR interne et externe. Il est toutefois possible d’identifier une altération de la BHRe dans certaines présentations cliniques.
-
Dans les œdèmes vasogéniques, comme les occlusions veineuses rétiniennes, la présence de liquide sous-rétinien traduit une probable rupture associée de la BHRe. En effet, la libération de VEGF par la rétine ischémique altère la fonction de barrière de l’EPR via le récepteur VEGF-1 (Flt-1), dont l’expression est régulée par hypoxia inducible factor 1a (HIF-1a).
-
La choroïdopathie hypertensive se caractérise par de multiples zones d’occlusion choriocapillaire, provoquant une souffrance focale de l’EPR, une rupture de la BHRe et l’accumulation de liquide sous-rétinien.
-
Dans les OM d’origine inflammatoire, il existe des altérations focales de l’EPR qui résultent d’une altération de la BHRe sous la dépendance de nombreux médiateurs de l’immunité.
-
Au cours de la rétinopathie diabétique, se produit une accumulation intrarétinienne de cellules microgliales dont l’élimination est diminuée en raison d’altérations de la plasticité du cytosquelette des cellules de l’EPR et des mécanismes de trans-cytose des cellules microgliales à travers les cellules de l’EPR [23]. Leur activation contribue à la libération d’oxyde nitrique (NO), de TNF-α, de diverses interleukines et d’anti-VEGF qui participent à la formation de l’OM. L’EPR soumis à une hyperglycémie chronique et au stress oxydatif participe aussi à la libération de médiateurs de l’inflammation comme le VEGF via l’activating transcription factor 4 (ATF-4), IL-6, IL-8, TNF-α, MCP-1, de chémokines, de thrombosporine-1 et d’autres facteurs solubles. Comme mentionné plus haut, il existe également une modification des aquaporines dans l’EPR ainsi qu’une diminution de l’occludine dans l’EPR (fig. 4-7) et la MLE (fig. 4-8).
-
Dans la choriorétinopathie séreuse centrale (CRSC), l’accumulation de liquide sous-rétinien résulte d’une altération focale de l’EPR classiquement dénommée « point de fuite » en raison de son apparence angiographique. Il s’agit d’un des rares cas d’altération évidente de la BHRe (fig. 4-9). Dans les formes chroniques de CRSC, l’atteinte étendue de l’épithélium pigmentaire, constatée par tomographie à cohérence optique et autofluorescence, pourrait expliquer la persistance du liquide sous-rétinien et l’œdème intrarétinien qui peut s’y associer. Il existe aussi probablement une altération des transports de fluide et d’électrolytes par l’EPR.
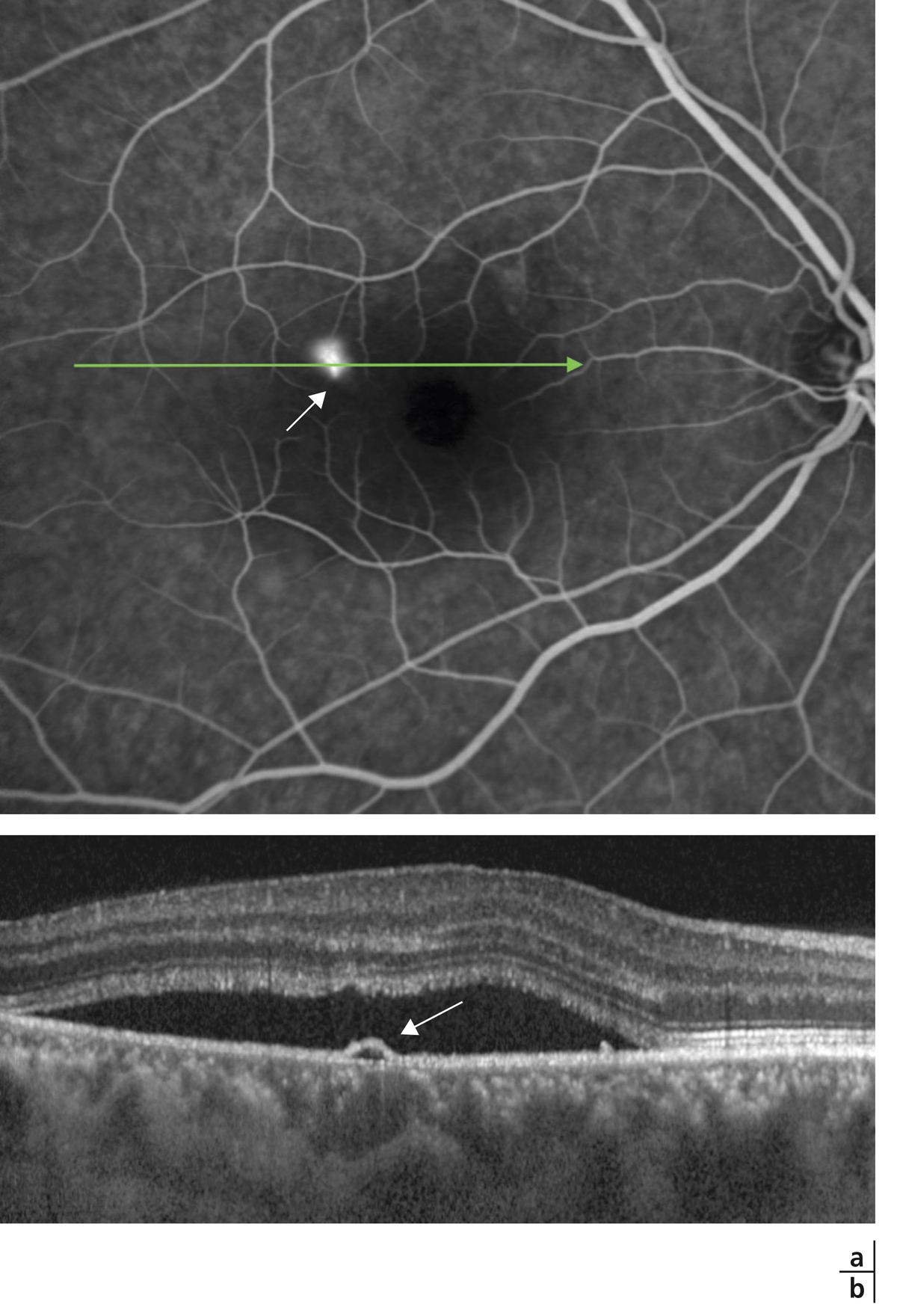
Fig. 4-9 Altération de la BHRe dans la choriorétinopathie séreuse centrale.
a. Angiographie à la fluorescéine chez un patient présentant un épisode aigu de choriorétinopathie séreuse centrale, montrant une altération focale de l’EPR révélée par un « point de fuite » (flèche). b. Section axiale en SD-OCT passant au niveau du « point de fuite » chez le même patient, montrant un décollement localisé de l’épithélium pigmentaire (flèche) et l’accumulation secondaire de liquide sous-rétinien.
Les œdèmes maculaires résultent de mécanismes intriqués impliquant des altérations des BHR interne et externe. La BHRe formée par les jonctions serrées et adhérentes entre les cellules de l’EPR joue un rôle critique dans l’équilibre hydro-ionique du tissu rétinien. Des données récentes indiquent que la membrane limitante externe participerait également à cette fonction de barrière. Diverses situations pathologiques associées à une altération de ces structures conduisent à un œdème maculaire. La compréhension précise des mécanismes impliqués permettra de mieux cibler les traitements disponibles et d’identifier de nouveaux agents pharmacologiques pour le traitement des œdèmes maculaires.
[1] Rizzolo LJ, Peng S, Luo Y, Xiao W. Integration of tight junctions and claudins with the barrier functions of the retinal pigment epithelium. Prog Retin Eye Res 2011 ; 30 : 296-323.
[2] Reichhart N, Strauss O. Ion channels and transporters of the retinal pigment epithelium. Exp Eye Res 2014 ; 126 : 27-37.
[3] Panda-Jonas S, Jonas JB, Jakobczyk-Zmija M. Retinal pigment epithelial cell count, distribution, and correlations in normal human eyes. Am J Ophthalmol 1996 ; 121 : 181-9.
[4] Finnemann SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E. Phagocytosis of rod outer segments by retinal pigment epithelial cells requires alpha(v)beta5 integrin for binding but not for internalization. Proc Natl Acad Sci USA 1997 ; 94 : 12932-7.
[5] Ryeom SW, Sparrow JR, Silverstein RL. CD36 participates in the phagocytosis of rod outer segments by retinal pigment epithelium. J Cell Sci 1996 ; 109 : 387-95.
[6] Tarnowski BI, Shepherd VL, Mclaughlin BJ. Mannose 6-phosphate receptors on the plasma membrane on rat retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1988 ; 29 : 291-7.
[7] Philp NJ, Wang D, Yoon H, Hjelmeland LM. Polarized expression of monocarboxylate transporters in human retinal pigment epithelium and ARPE-19 cells. Invest Ophthalmol Vis Sci 2003 ; 44 : 1716-21.
[8] Omri S, Omri B, Savoldelli M, et al. The outer limiting membrane (OLM) revisited : clinical implications. Clin Ophthalmol 2010 ; 4 : 183-95.
[9] Schlötzer-Schrehardt U, Müller HG, Wirtz PM, Naumann GO. Desmosomal-mitochondrial complexes in human nonpigmented ciliary and retinal pigment epithelia. Invest Ophthalmol Vis Sci 1990 ; 31 : 664-9.
[10] Asayama K. In vivo study on the absorption of the subretianl fluid. 2. Studies on an absorption of tracers (I125.human serum albumin and lanthanum nitrate) injected between the sensory retina and the pigment epithelium layer. Nippon Ganka Gakkai Zasshi 1976 ; 80 : 598-607.
[11] Bringmann A, Pannicke T, Grosche J, et al. Müller cells in the healthy and diseased retina. Prog Retin Eye Res 2006 ; 25 : 397-424.
[12] Wimmers S, Karl MO, Strauss O. Ion channels in the RPE. Prog Retin Eye Res 2007 ; 26 : 263-301.
[13] Bialek S, Miller SS. K+ and Cl– transport mechanisms in bovine pigment epithelium that could modulate subretinal space volume and composition. J Physiol 1994 ; 475 : 401-17.
[14] Tsuboi S, Pederson JE. Effect of plasma osmolality and intraocular pressure on fluid movement across the blood-retinal barrier. Invest Ophthalmol Vis Sci 1988 ; 29 : 1747-9.
[15] Tsuboi S, Pederson JE. Volume flow across the isolated retinal pigment epithelium of cynomolgus monkey eyes. Invest Ophthalmol Vis Sci 1988 ; 29 : 1652-5.
[16] Verkman AS, Ruiz-Ederra J, Levin MH. Functions of aquaporins in the eye. Prog Retin Eye Res 2008 ; 27 : 420-33.
[17] Rosenthal R, Heimann H, Agostini H, et al. Ca2+ channels in retinal pigment epithelial cells regulate vascular endothelial growth factor secretion rates in health and disease. Mol Vis 2007 ; 13 : 443-56.
[18] Hollborn M, Dukic-Stefanovic S, Pannicke T, et al. Expression of aquaporins in the retina of diabetic rats. Curr Eye Res 2011 ; 36 : 850-6.
[19] Omri S, Behar-Cohen F, Rothschild PR, et al. PKCζ mediates breakdown of outer blood-retinal barriers in diabetic retinopathy. PLoS ONE 2013 ; 8 : e81600.
[20] De Bock M, Wang N, Decrock E, et al. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog Neurobiol 2013 ; 108 : 1-20.
[21] Alexander JS, Elrod JW. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J Anat 2002 ; 200 : 561-74.
[22] Jain A, Saxena S, Khanna VK, et al. Status of serum VEGF and ICAM-1 and its association with external limiting membrane and inner segment-outer segment junction disruption in type 2 diabetes mellitus. Mol Vis 2013 ; 19 : 1760-8.
[23] Omri S, Behar-Cohen F, De Kozak Y, et al. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy : role of PKCζ in the Goto Kakizaki rat model. Am J Pathol 2011 ; 179 : 942-53.
A. Giocanti-Aurégan
➤ Les cellules gliales de Müller sont responsables du maintien de l’homéostasie rétinienne indispensable au bon fonctionnement des neurones rétiniens et de l’élimination des neurotransmetteurs en excès.
➤ Pour cela, elles possèdent des canaux aqueux (AQP4) et potassiques (Kir4.1), permettant d’absorber et d’éliminer du fluide et du potassium et de maintenir ainsi l’équilibre osmotique de la rétine. Elles possèdent également des transporteurs permettant la recapture de neurotransmetteurs et leur recyclage.
➤ En conditions pathologiques (œdème maculaire), ces capacités sont dépassées. Ni l’homéostasie, ni l’élimination des neurotransmetteurs ne sont assurées. Ces phénomènes sont potentiellement toxiques pour les neurones.
Cellules gliales de la rétine : cellules situées dans la rétine qui entourent les neurones et participent au contrôle de l’environnement chimique et électrique, en leur fournissant des nutriments et en éliminant leurs déchets. Il existe trois types de cellules gliales : les astrocytes rétiniens, les cellules gliales de Müller (ces deux types cellulaires étant appelés cellules macrogliales) et les cellules microgliales (non abordées dans ce sous-chapitre).
Homéostasie rétinienne : l’homéostasie est un phénomène par lequel un facteur clé est maintenu autour d’une valeur bénéfique pour le système considéré par un processus de régulation. Dans la rétine, l’homéostasie concerne le maintien des conditions nécessaires (pH, osmolarité) permettant un fonctionnement neuronal normal.
Canaux potassiques Kir4.1 : canaux transmembranaires, localisés sur la membrane plasmatique des cellules gliales de Müller, responsables de la réabsorption et de l’élimination vers le vitré et les vaisseaux sanguins de potassium, permettant l’équilibre ionique du milieu extracellulaire.
Canaux aqueux AQP4 : canaux transmembranaires, localisés sur la membrane plasmatique des cellules gliales de Müller, responsables de la réabsorption et de l’élimination vers le vitré et les vaisseaux sanguins de fluide, permettant la réabsorption rapide des fluides intrarétiniens.
La rétine des mammifères contient trois types de cellules gliales. Outre les cellules microgliales, il existe deux types de cellules macrogliales qui assurent un soutien aux cellules neuronales : les astrocytes et les cellules gliales de Müller (CGM). Les astrocytes et les CGM sont en contact direct avec le plexus vasculaire superficiel rétinien et avec l’interface vitréorétinien via des extensions cytoplasmiques. Les CGM sont particulièrement impliquées dans les mécanismes d’homéostasie et de cohésion de la rétine [1].
La cellule gliale de Müller (CGM) est la principale cellule gliale de la rétine des vertébrés. Les CGM sont des cellules gliales radiales spécialisées qui s’étendent sur toute l’épaisseur de la rétine (fig. 4-10) et sont en contact avec tous les corps cellulaires des neurones de la rétine.
Ainsi, les CGM constituent un lien anatomique entre les neurones rétiniens et les compartiments (les vaisseaux sanguins rétiniens, le vitré, et l’espace sous-rétinien) avec lesquels elles échangent des molécules. Ce lien n’est pas seulement anatomique mais aussi fonctionnel. À cet effet, les CGM sont dotées de différents canaux ioniques, de récepteurs de ligand, de protéines de transport transmembranaires et d’enzymes [2]. Nombre de ces molécules sont spécifiquement exprimées par les CGM. Une caractéristique clé des CGM normales est la conductance élevée en potassium (K+) de leur membrane plasmatique. Celle-ci est assurée grâce à la forte densité de canaux K+ spécialisés dans la réabsorption du potassium extracellulaire vers l’intérieur de la CGM, puis l’élimination de ce potassium vers les capillaires rétiniens et le vitré [3].
Dans la rétine saine, les CGM (fig. 4-11) :
-
sont impliquées dans le métabolisme du glucose, fournissent les neurones en nutriments tels que lactate/pyruvate pour leur métabolisme oxydatif [4, 5] et favorisent l’élimination des déchets, produits par leur métabolisme ;
-
régulent le flux sanguin rétinien [6] et contribuent à la formation et au maintien de la BHR par leur proximité avec les vaisseaux rétiniens [7] ;
-
contribuent au processus de signalisation neuronale, particulièrement par recapture immédiate et recyclage des neurotransmetteurs [8] et fournissent les précurseurs des neurotransmetteurs aux neurones ;
-
maintiennent l’homéostasie aqueuse et ionique de la rétine et ainsi le pH [1, 2] ;
-
libèrent des facteurs (D-serine et glutamate) qui contrôlent l’excitabilité des neurones [9, 10].
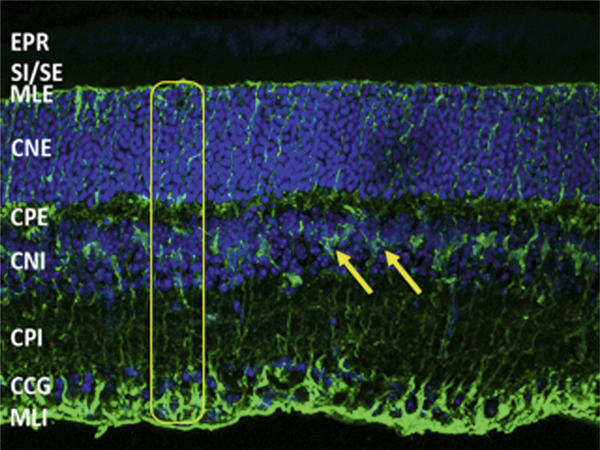
Fig. 4-10 Coupe de rétine de souris marquée pour la glutamine synthétase (vert) marqueur des cellules gliales de Müller, et DAPI (bleu), marqueur des noyaux cellulaires.
La cellule gliale de Müller (entourée en jaune) s’étend au travers des différentes couches rétiniennes de la limitante interne jusqu’à la nucléaire externe. Les corps cellulaires (flèches jaunes) sont bien visibles et ont des prolongements vers la rétine interne et externe. CCG : couche cellules ganglionnaires ; CNE : couche nucléaire externe ; CNI : couche nucléaire interne ; CPE : couche plexiforme externe ; CPI : couche plexiforme interne ; EPR : épithélium pigmentaire rétinien ; MLE : membrane limitante externe ; MLI : membrane limitante interne ; SI/SE : jonction des segments internes et externes des photorécepteurs.
(Source : Dr Ophélie Vaccan, PhD, Institut de la vision.)
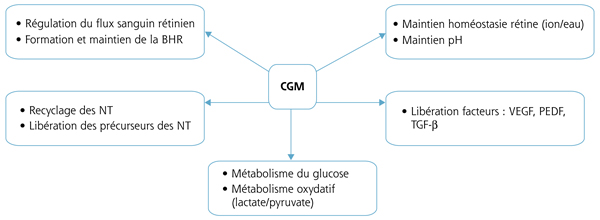
Fig. 4-11 Résumé des différentes fonctions connues de la cellule gliale de Müller (CGM) [1].
BHR : barrière hémato-rétinienne ; NT : neurotransmetteurs ; PEDF : pigment epithelium derived factor ; TGF : transforming growth factor ; VEGF : vascular endothelial growth factor.
Dès les stades embryonnaires précoces, la CGM, même immature, est importante pour l’organisation de la rétine en développement et la mise en place des circuits neuronaux. Durant les premiers jours après la naissance, les cellules gliales radiales immatures ont une disposition caractéristique, servant de support à l’activité mitotique des cellules. Les CGM ne sont pas encore dotées de capacités de courant K+ entrant. En revanche, dans les CGM différenciées, au-delà de la deuxième semaine de vie, une augmentation des courants entrants de K+ se produit. Elle est responsable d’une hyperpolarisation du potentiel de membrane, comparable à celle de l’adulte (de l’ordre de –80 mV) [11–13] grâce au positionnement de canaux Kir4.1 au niveau de la membrane des CGM [3]. Cette homéostasie potassique assurée par les CGM matures est essentielle pour maintenir un fonctionnement neuronal correct. En parallèle, les jeunes CGM sont impliquées dans le guidage du développement vasculaire d’une partie des vaisseaux rétiniens [14] ainsi que dans l’induction de la BHR [7].
Les CGM, contrairement aux neurones rétiniens, sont étonnamment résistantes à une grande variété de facteurs pathogènes, y compris l’ischémie, l’hypoxie et l’hypoglycémie. Une raison de leur insensibilité à ce type de traumatismes est leur métabolisme énergétique spécialisé. Les CGM utilisent essentiellement la glycolyse anaérobie, même en présence d’oxygène. Par conséquent, elles affichent une faible consommation d’oxygène et peuvent résister à une longue durée d’anoxie [15, 16].
Tant que l’oxygène est disponible, les CGM résistent également à l’absence de glucose si d’autres substrats tels que le lactate, le pyruvate, le glutamate, la glutamine peuvent être métabolisés pour produire des substrats énergétiques [16, 17]. Ces mécanismes impactent l’interaction entre CGM et neurones rétiniens. En effet, le métabolisme glycolytique des CGM produit du lactate, qui est converti en pyruvate par les enzymes gliales. Le pyruvate est ensuite libéré par les CGM et capté par les neurones qui l’utilisent comme un substrat pour leur propre cycle de Krebs (fig. 4-12) [15]. L’anhydrase carbonique, enzyme spécifique de la glie, permet de convertir le dioxyde de carbone (CO2) qui est toxique en ions bicarbonates qui sont éliminés vers le vitré ou vers les vaisseaux sanguins (voir fig. 4-12).
Ainsi, les CGM sont responsables de l’élimination de produits de dégradation potentiellement toxiques pour la rétine (les neurones sont incapables d’inactiver le CO2 eux-mêmes). Ce type d’interaction est appelé « symbiose métabolique » entre les neurones rétiniens et les cellules gliales (voir fig. 4-12).
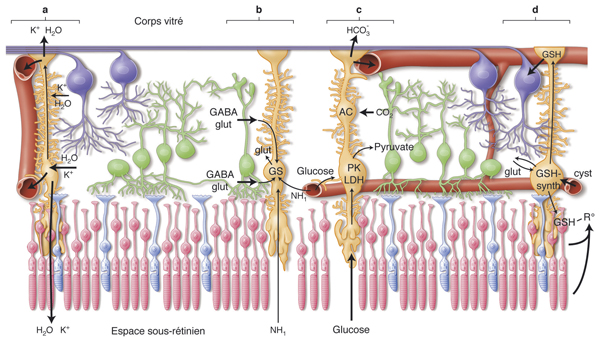
Fig. 4-12 Interactions entre neurones et cellule gliale de Müller dans une rétine normale.
a. Tamponnement des ions K+. b. Recyclage des neurotransmetteurs. c. Symbiose métabolique. d. Élimination des radicaux libres. AC : anhydrase carbonique ; cyst : cystéine ; GABA : acide gamma-aminobutyrique ; glut : glutamate ; GS : glutamine synthétase ; GSH : glutathion ; LDH : lactate déshydrogénase ; PK : pyruvate kinase ; R° : radicaux libres.
(D’après Bringmann A, et al. [1].)
Les CGM jouent un rôle majeur dans le recyclage des neurotransmetteurs. Les CGM permettent l’élimination rapide des neurotransmetteurs des espaces synaptiques. Pour cela, elles expriment des systèmes d’absorption des acides aminés tels que le glutamate, l’acide γ-aminobutyrique (GABA), et la glycine [1]. La clairance du glutamate synaptique par les CGM est nécessaire pour le fonctionnement normal des synapses excitatrices et pour la prévention de la neurotoxicité [18].
Les CGM capturent le glutamate et le convertissent en glutamine grâce à l’enzyme glutamine synthétase (GS) qui n’est exprimée que dans les CGM. La glutamine est transportée vers les neurones qui l’utilisent comme précurseur pour la synthèse de nouveaux neurotransmetteurs comme le glutamate et le GABA [19]. Lorsque la GS est expérimentalement bloquée dans les CGM, les neurones rétiniens perdent leur teneur en glutamate et les animaux deviennent aveugles [20, 21].
Les CGM sont fondamentalement impliquées dans l’homéostasie rétinienne des ions K+, en assurant un transfert transmembranaire de cet ion. Cette fonction permet d’équilibrer les modifications de concentrations potassiques extracellulaires associées à l’activité neuronale [2, 22]. Les neurones libèrent activement des ions K+ en particulier au niveau des couches plexiformes (correspondant à la localisation des synapses). Les CGM captent l’excès de K+ dérivé des neurones à ce niveau et éliminent une quantité similaire de K+ dans des espaces liquidiens en dehors de la rétine neuronale (sang, vitré et espace sous-rétinien). Il existe divers mécanismes d’entrée du K+ dans les CGM. Le canal Kir4.1 est responsable des courants entrants rectificateurs de K+ [1, 3].
Il existe une polarisation de l’expression des canaux Kir4.1 dans la membrane plasmatique des CGM, avec une forte densité de ces canaux dans les domaines plasmatiques en contact avec les zones d’excès de K+ extracellulaire, dans les espaces périvasculaires, et au niveau de la membrane limitante interne au contact du vitré (voir fig. 4-12), en contact avec les zones d’élimination du K+ [3, 23].
En cas de pathologies rétiniennes, les canaux Kir4.1 peuvent être délocalisés ou moins nombreux et perdent ainsi leur polarisation [13]. Un dérèglement de l’homéostasie du K+ peut être à l’origine d’une hyperexcitation neuronale et d’une excitotoxicité (toxicité liée au glutamate).
Dans des conditions normales, l’eau s’accumule dans le tissu rétinien, provenant :
-
de la production endogène d’eau associée à la synthèse oxydative de l’adénosine 5'-triphosphate (ATP) ;
-
d’un afflux d’eau provenant de la circulation sanguine couplé à la capture de substrats métaboliques tels que le glucose ;
-
d’un passage d’eau dans la rétine par élévation de la pression intra-oculaire [24].
Cette eau est éliminée en permanence par l’épithélium pigmentaire (EP) qui déshydrate l’espace sous-rétinien, et par les CGM qui déshydratent la rétine interne [1]. L’EP et les CGM permettent l’élimination d’eau par des flux transcellulaires aqueux grâce à des canaux aqueux de type aquaporines. Les CGM permettent l’élimination aqueuse du milieu interstitiel vers l’intérieur des cellules puis vers les vaisseaux sanguins (fig. 4-13). Le transport d’eau par les canaux AQP4 au niveau des CGM suit le flux entrant de K+ par les canaux Kir4.1 [23]. La co-expression des AQP4 et Kir4.1 suggère que les différences osmotiques entre le tissu rétinien et le sang ou vitré sont compensées par les entrées et sorties d’eau et de K+ des CGM en fonction de l’activité neuronale du moment.
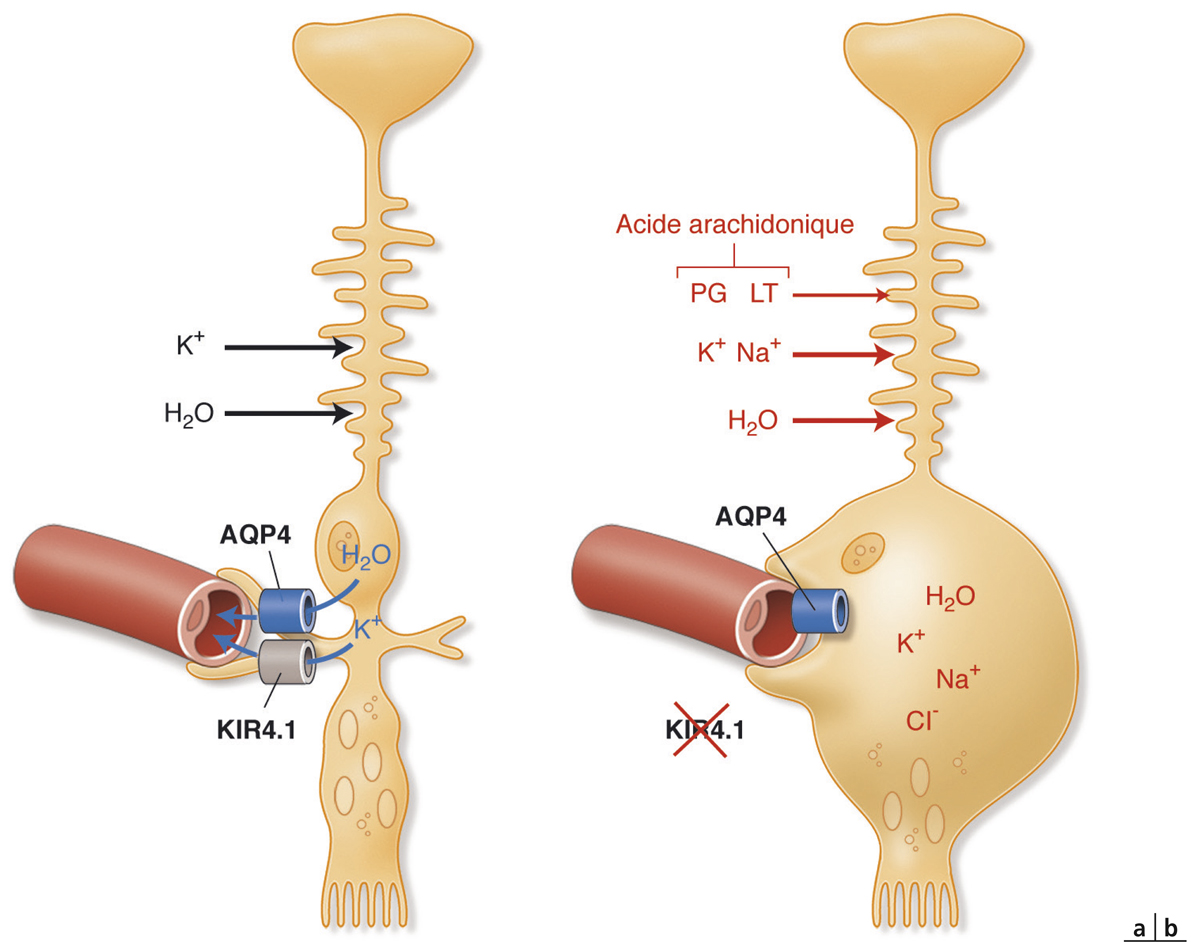
Fig. 4-13 Ballonisation des cellules gliales de Müller (CGM) en conditions pathologiques.
a. Dans la rétine normale, les CGM déshydratent la rétine et réabsorbent l’eau et K+ du milieu extracellulaire (libéré par les neurones activés) puis les éliminent vers la circulation sanguine. b. Les CGM de rétines altérées par l’ischémie, l’inflammation ou le diabète, diminuent l’expression des canaux Kir4.1 exprimés au niveau des zones périvasculaires de la membrane plasmatique des CGM, ce qui diminue l’élimination des ions K+ vers le sang et augmente la pression osmotique à l’intérieur des CGM et conduit à une ballonisation de ces cellules. PG : prostaglandines ; LT : leucotriènes.
(D’après Bringmann A, et al. [1].)
La présence d’un œdème maculaire (OM) est souvent responsable d’une baisse d’acuité visuelle [25]. L’œdème peut contribuer à la dégénérescence des photorécepteurs, à la mort des cellules neuronales par compression des fibres nerveuses, des neurones et des capillaires rétiniens. Le développement d’un OM dépend de deux paramètres : le flux de liquide entrant dans le parenchyme rétinien à travers les parois des vaisseaux devenus perméables ; le taux de réabsorption de fluide à partir du tissu rétinien vers la circulation sanguine par l’EP et les CGM. L’OM peut être causé par une rupture de la BHRi (fuites vasculaires provoquant un œdème extracellulaire) et/ou par un gonflement des CGM (œdème intracellulaire). Il a été montré que l’œdème maculaire diabétique se produit uniquement lorsqu’il existe une perturbation de la réabsorption du fluide rétinien en plus de la fuite vasculaire. Le gonflement des CGM semble également impliqué dans le développement de l’OM cystoïde, dans lequel les logettes kystiques souvent visibles en angiographie correspondraient aux CGM ballonisées [26–28]. Le transport d’eau au travers des CGM est couplé à des courants K+. En conditions pathologiques d’œdème maculaire, il semblerait qu’il existe une délocalisation des canaux aqueux et potassiques ne permettant plus l’élimination d’eau et de potassium de l’intérieur des CGM vers les vaisseaux rétiniens et le vitré entraînant un gonflement des CGM [1].
Les corticoïdes, en plus de leur effet dans le rétablissement des jonctions serrées entre les cellules endothéliales permettant une restauration de la BHRi (voir chapitre 4.2), peuvent agir sur ces phénomènes de transport aqueux et ioniques. En effet, la dexaméthasone et la triamcinolone permettent de modifier la localisation et le nombre des canaux aqueux et potassiques [29].
Dans une rétine saine, la CGM participe au maintien de l’homéostasie rétinienne grâce à l’élimination rapide des fluides vers les vaisseaux rétiniens et le vitré.
L’absorption et l’élimination des fluides par les CGM sont assurées par des canaux aqueux AQP4 et potassiques Kir4.1.
Dans des conditions pathologiques et sous l’influence de facteurs inflammatoires, les canaux potassiques et aqueux transmembranaires des CGM sont altérés, ce qui diminue l’élimination de fluide et de potassium vers la circulation générale et contribue à une ballonisation des CGM (part intracellulaire de l’œdème rétinien).
[1] Bringmann A, Pannicke T, Grosche J, et al. Müller cells in the healthy and diseased retina. Prog Retin Eye Res 2006 ; 25 : 397-424.
[2] Newman E, Reichenbach A. The Müller cell : a functional element of the retina. Trends Neurosci 1996 ; 19 : 307-12.
[3] Kofuji P, Biedermann B, Siddharthan V, et al. Kir potassium channel subunit expression in retinal glial cells : implications for spatial potassium buffering. Glia 2002 ; 39 : 292-303.
[4] Poitry-Yamate CL, Poitry S, Tsacopoulos M. Lactate released by Müller glial cells is metabolized by photoreceptors from mammalian retina. J Neurosci Off J Soc Neurosci 1995 ; 15 : 5179-91.
[5] Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci Off J Soc Neurosci 1996 ; 16 : 877-85.
[6] Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow ? Science 1987 ; 237 : 896-8.
[7] Tout S, Chan-Ling T, Holländer H, Stone J. The role of Müller cells in the formation of the blood-retinal barrier. Neuroscience 1993 ; 55 : 291-301.
[8] Matsui K, Hosoi N, Tachibana M. Active role of glutamate uptake in the synaptic transmission from retinal nonspiking neurons. J Neurosci Off J Soc Neurosci 1999 ; 19 : 6755-66.
[9] Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci Off J Soc Neurosci 1998 ; 18 : 4022-8.
[10] Stevens ER, Esguerra M, Kim PM, et al. D-serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc Natl Acad Sci 2003 ; 100 : 6789-94.
[11] Bringmann A, Biedermann B, Reichenbach A. Expression of potassium channels during postnatal differentiation of rabbit Müller glial cells. Eur J Neurosci 1999 ; 11 : 2883-96.
[12] Felmy F, Pannicke T, Richt JA, et al. Electrophysiological properties of rat retinal Müller (glial) cells in postnatally developing and in pathologically altered retinae. Glia 2001 ; 34 : 190-9.
[13] Pannicke T, Fischer W, Biedermann B, et al. P2X7 receptors in Müller glial cells from the human retina. J Neurosci Off J Soc Neurosci 2000 ; 20 : 5965-72.
[14] Wolburg H, Liebner S, Reichenbach A, Gerhardt H. The pecten oculi of the chicken : a model system for vascular differentiation and barrier maturation. Int Rev Cytol 1999 ; 187 : 111-59.
[15] Poitry-Yamate CL, Poitry S, Tsacopoulos M. Lactate released by Müller glial cells is metabolized by photoreceptors from mammalian retina. J Neurosci Off J Soc Neurosci 1995 ; 15 : 5179-91.
[16] Winkler BS, Arnold MJ, Brassell MA, Puro DG. Energy metabolism in human retinal Müller cells. Invest Ophthalmol Vis Sci 2000 ; 41 : 3183-90.
[17] Tsacopoulos M, Poitry-Yamate CL, Macleish PR, Poitry S. Trafficking of molecules and metabolic signals in the retina. Prog Retin Eye Res 1998 ; 17 : 429-42.
[18] Barnett NL, Pow DV. Antisense knockdown of GLAST, a glial glutamate transporter, compromises retinal function. Invest Ophthalmo Vis Sci 2000 ; 41 : 585-91.
[19] Pow DV, Crook DK. Direct immunocytochemical evidence for the transfer of glutamine from glial cells to neurons : use of specific antibodies directed against the d-stereoisomers of glutamate and glutamine. Neuroscience 1996 ; 70 : 295-302.
[20] Barnett NL, Pow DV, Robinson SR. Inhibition of Müller cell glutamine synthetase rapidly impairs the retinal response to light. Glia 2000 ; 30 : 64-73.
[21] Pow DV, Robinson SR. Glutamate in some retinal neurons is derived solely from glia. Neuroscience 1994 ; 60 : 355-66.
[22] Reichenbach A, Henke A, Eberhardt W, et al. K+ ion regulation in retina. Can J Physiol Pharmacol 1992 ; 70 Suppl : S239-247.
[23] Nagelhus EA, Veruki ML, Torp R, et al. Aquaporin-4 water channel protein in the rat retina and optic nerve : polarized expression in Müller cells and fibrous astrocytes. J Neurosci Off J Soc Neurosci 1998 ; 18 : 2506-19.
[24] Marmor MF. Mechanisms of fluid accumulation in retinal edema. Doc Ophthalmol Adv Ophthalmol 1999 ; 97 : 239-49.
[25] Larsen M, Wang M, Sander B. Overnight thickness variation in diabetic macular edema. Invest Ophthalmo Vis Sci 2005 ; 46 : 2313-6.
[26] Bringmann A, Reichenbach A, Wiedemann P. Pathomechanisms of cystoid macular edema Ophthalmic Res 2004 ; 36 : 241-9.
[27] Fine BS, Brucker AJ. Macular edema and cystoid macular edema. Am J Ophthalmol 1981 ; 92 : 466-81.
[28] Yanoff M, Fine BS, Brucker AJ, Eagle RC. Pathology of human cystoid macular edema. Surv Ophthalmol 1984 ; 28 Suppl : 505-11.
[29] Zhao M, Bousquet E, Valamanesh F, et al. Differential regulations of AQP4 and Kir4.1 by triamcinolone acetonide and dexamethasone in the healthy and inflamed retina. Invest Ophthalmo Vis Sci 2011 ; 52 : 6340-7.
B. Dupas
➤ Dans l’œdème maculaire, l’accumulation de liquide est à la fois intra- et extracellulaire.
➤ La baisse visuelle est liée à la diffraction des rayons lumineux à travers la rétine œdémateuse et également à l’altération neuronale et gliale induite par les modifications de l’homéostasie rétinienne secondaires à l’œdème.
➤ Un œdème rétinien récent et traité rapidement (comme un syndrome d’Irvine-Gass) ne laisse en général pas de séquelles rétiniennes.
➤ Les œdèmes des occlusions veineuses induisent des remodelages microvasculaires à l’origine d’œdèmes chroniques.
➤ L’altération neurogliale dans le cadre d’un œdème maculaire diabétique chronique est liée à l’œdème, mais également à une perte des cellules ganglionnaires inhérentes à la neuropathie diabétique.
➤ La récupération visuelle dépend du degré de perte neurogliale induite par l’œdème maculaire, souvent lié à la durée d’évolution de la maladie causale.
La perte de l’homéostasie rétinienne induite par la rupture des BHR interne et externe conduit à des altérations fonctionnelles d’abord réversibles, puis organiques irréversibles lorsque l’œdème maculaire (OM) devient chronique. Les anomalies morphologiques rétiniennes sont beaucoup mieux identifiables une fois que l’OM est résorbé, car l’analyse optical coherence tomography (OCT) des structures rétiniennes en coupes horizontales ou verticales à la phase œdémateuse est rendue difficile par de nombreux artefacts. Cependant l’avènement des systèmes d’imagerie « en face » et de l’OCT-angiographie permet un meilleur phénotypage de la maladie.
L’OM, si on le définit par une accumulation de fluide dans la rétine, débuterait dans le compartiment intracellulaire. Les cellules gliales de Müller (CGM) semblent les premières cellules affectées. Des études en microscopie électronique sur des yeux atteints d’OM cystoïde diabétique [1] révèlent une ballonisation du cytoplasme cellulaire avec une préservation et un respect de la taille des espaces intercellullaires (voir fig. 2-13 et 2-14). Le processus œdémateux semble débuter par un gonflement intracytoplasmique des cellules de Müller. Cet œdème progressif mène à la nécrose de la cellule et s’accompagne de dégénérescence neuronale. Ces deux phénomènes conduisent à la formation de larges « kystes ». Cependant histologiquement les logettes observées à l’angiographie ne sont pas retrouvées. En effet, il semblerait, aux stades précoces, qu’il n’existe pas d’élargissement de l’espace intercellulaire, et l’accumulation de fluorescéine aurait lieu dans le cytoplasme des cellules de Müller. La paroi des kystes serait composée de cellules compactées, pouvant correspondre à des CGM gardant leur attache par des desmosomes. Les cellules neuronales (bipolaires, ganglionnaires et photorécepteurs) sont également victimes de modifications morphologiques, avec notamment un élargissement de l’appareil présynaptique et une diminution de leurs prolongements. Le segment externe des photorécepteurs est quant à lui le plus souvent intègre. À ce stade, l’épaisseur rétinienne peut rester normale, avec pourtant une rupture de la BHRi matérialisable par des diffusions de fluorescéine [2], comme cela a été montré dans les premiers stades de rétinopathie diabétique chez des patients indemnes de toute anomalie vasculaire décelable au fond d’œil (fig. 4-14).
Ce dysfonctionnement primitif, à l’origine d’une modification de l’homéostasie et des flux transrétiniens, aboutit rapidement à l’accumulation de liquide dans l’espace extracellulaire, avec épaississement rétinien cliniquement détectable [3]. L’œdème semble réversible avec un potentiel de récupération essentiellement au niveau des CGM, alors que les cellules neuronales sont beaucoup plus vulnérables. Le mécanisme de vacuolisation des CGM n’est pas clair mais pourrait provenir soit de l’atteinte primitive métabolique des CGM, soit des anomalies vasculaires (dont la rupture de la BHR), soit d’une déficience de l’épithélium pigmentaire rétinien (RPE), ou plus probablement d’un mécanisme associant les trois éléments. La présence de liquide va physiquement modifier les relations entre les neurones et les cellules gliales, tout en majorant leur dysfonctionnement, aggravant la rupture de la BHR, créant ainsi un cercle vicieux en partie médié par des cytokines inflammatoires. Le liquide intrarétinien accumulé, traversé de « piliers cellulaires » constitués de cellules bipolaires et de cellules de Müller compactées [4], apparaît en coupe OCT comme des « kystes » (fig. 4-15 et 4-16). Si le liquide semble s’accumuler entre les couches plexiformes externes et internes, la disposition peut varier selon l’étiologie de l’OM :
-
l’OM diabétique (OMD) est d’apparition progressive. Il débute souvent dans les couches internes, car il est secondaire à des diffusions à partir de micro-anévrismes du lit capillaire (fig. 4-15). L’OCT-angiographie permet une analyse précise de ces anomalies observées dans les lits capillaires superficiels et profonds (fig. 4-16) ;
-
l’OM des occlusions veineuses rétiniennes (OVR) est quant à lui beaucoup plus brutal et secondaire à une hypoperfusion atteignant la totalité du lit capillaire, consécutif au ralentissement des flux circulatoires. L’œdème prédomine le plus souvent dans la couche nucléaire externe et ce phénomène serait lié au blocage des protéines de gros poids moléculaire par la membrane limitante externe [5] ;
-
un décollement séreux rétinien (DSR) peut fréquemment s’associer aux OM des OVR et du diabète (prévalence du DSR dans les OVR : 80 % versus dans l’OMD : 20 %). Ces DSR ne se remplissent pas de fluorescéine [6], ce qui laisse penser qu’ils proviendraient d’un dysfonctionnement de l’EP (défaut de réabsorption) plutôt que d’une hyperperméabilité choroïdienne [7]. Certains auteurs évoquent également la possibilité de rupture de kystes intrarétiniens qui se videraient dans l’espace sous-rétinien [8]. Cependant le DSR ne constitue pas un marqueur de mauvais pronostic visuel [9] ;
-
les OM inflammatoires des uvéites peuvent s’associer à des vascularites et l’angiographie en fluorescéine et au vert d’indocyanine (indocyanine green ou ICG) est indispensable dans le bilan initial de la maladie ;
-
enfin, à part, il faut citer les œdèmes des occlusions artérielles rétiniennes, qui se distinguent par l’absence d’exsudation intrarétinienne et par un épaississement rétinien ischémique global des couches internes (hyperréflectivité à l’OCT), suivi d’une atrophie secondaire de ces couches.
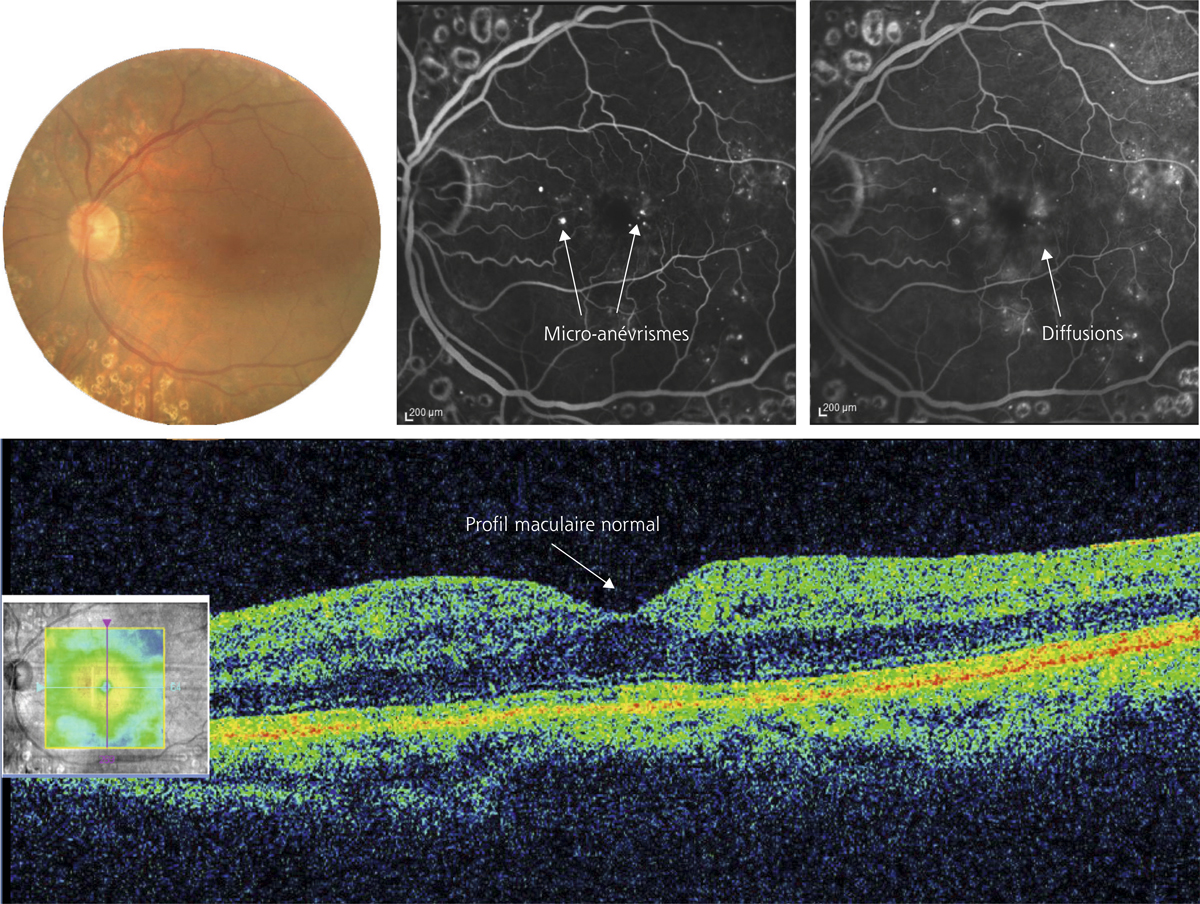
Fig. 4-14 Diffusion sans œdème.
Des diffusions de fluorescéine sont visibles à partir de micro-anévrismes, témoignant de la rupture de la BHR, alors qu’aucun épaississement rétinien n’est noté en OCT.
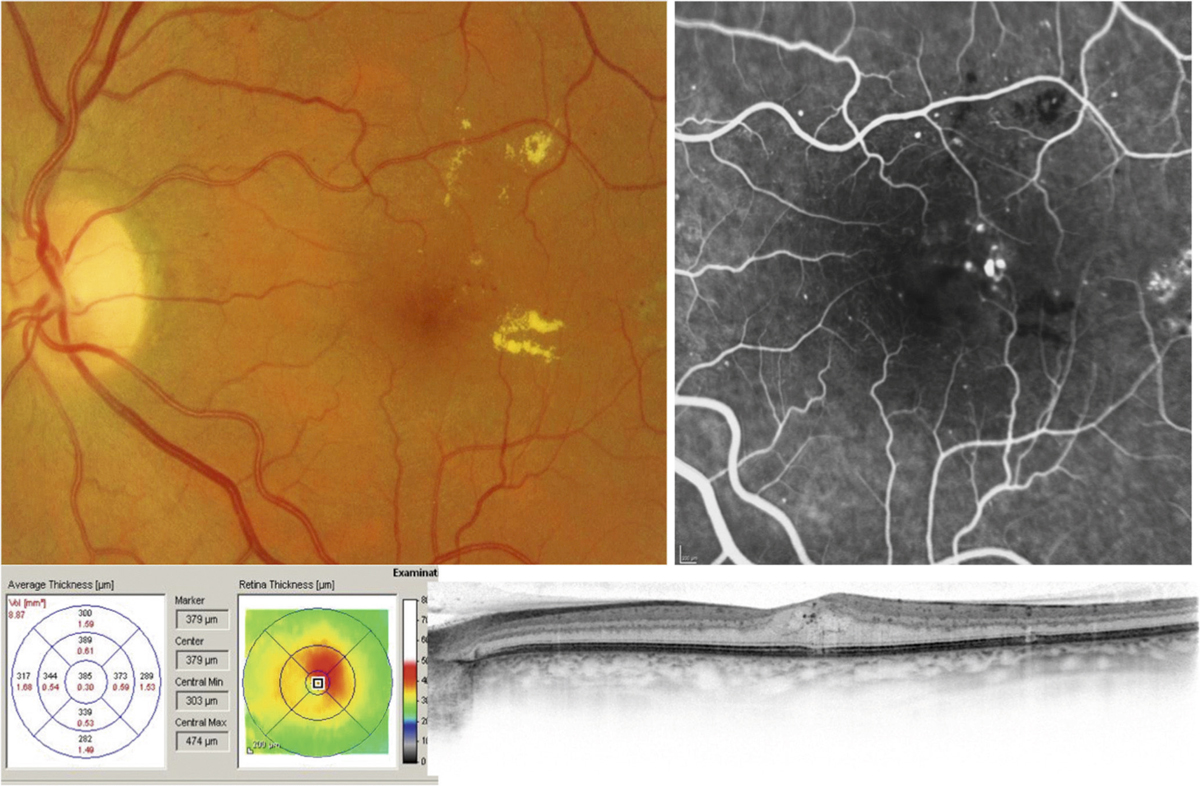
Fig. 4-15 Œdème maculaire diabétique débutant.
Les micro-anévrismes visibles en angiographie sont responsables d’un épaississement rétinien localisé, entouré d’exsudats. L’acuité visuelle est conservée à 10/10e.
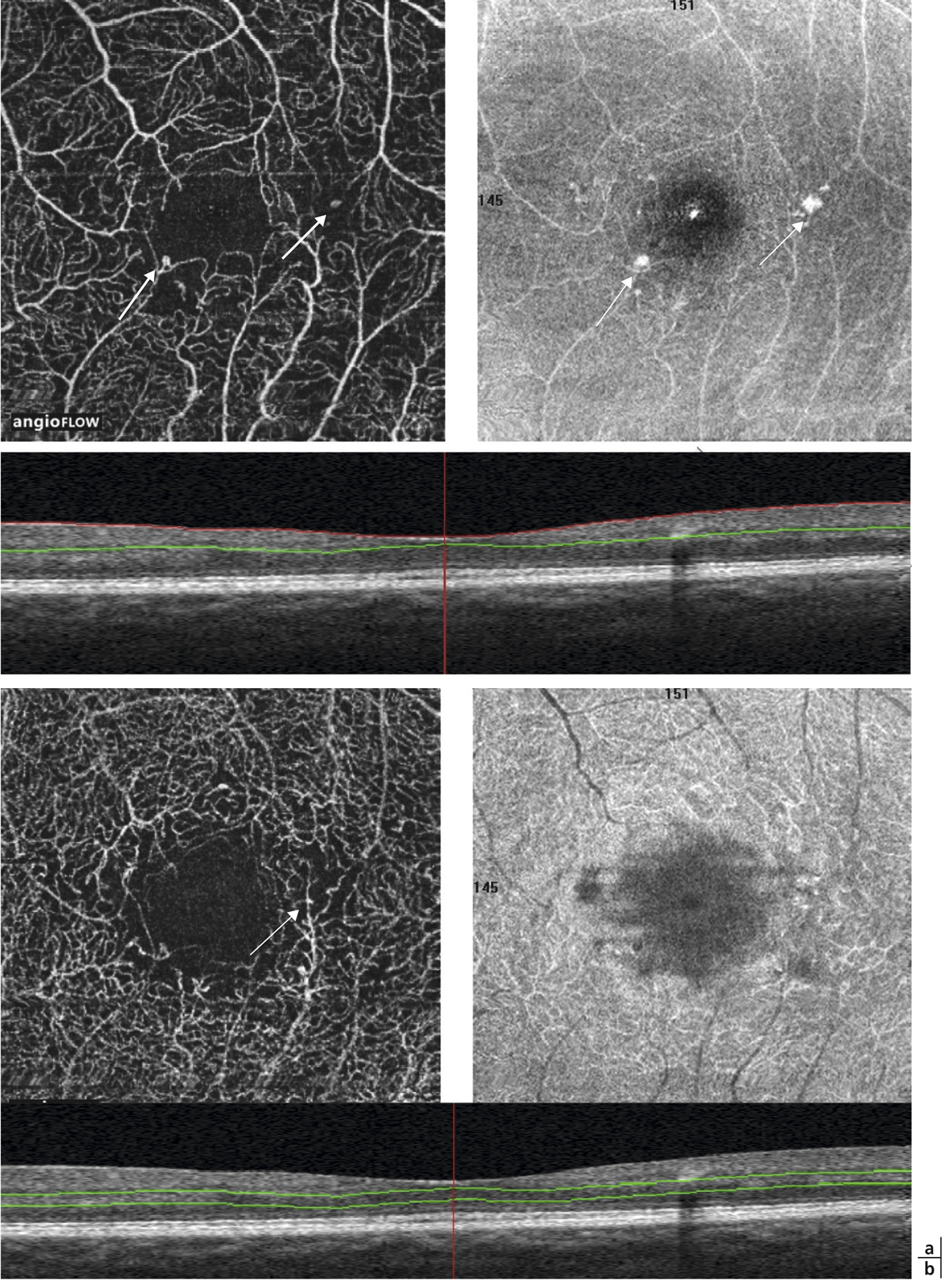
Fig. 4-16 Anomalies de la maille capillaire périfovéolaire chez un patient diabétique en OCT-angiographie.
a. Micro-anévrismes visibles dans le lit capillaire rétinien superficiel (flèche). b. Le lit capillaire profond révèle une architecture différente avec des capillaires raréfiés (flèche).
Si l’OM trouve majoritairement son origine dans la rupture de la BHRi, la totalité de la rétine (externe et interne) est affectée par la perte neuronale lorsque ce dernier se chronicise.
Dans l’OMD, la neuropathie diabétique contribue à générer une apoptose des cellules rétiniennes, notamment ganglionnaires [10] même en l’absence d’OM, qui s’ajoute à la perte cellulaire engendrée par ce dernier. C’est ainsi qu’après résolution de l’OMD, il est observé une diminution de l’épaisseur de la couche des cellules ganglionnaires rétiniennes [11].
Une prolifération gliale de surface est également fréquemment rencontrée après OVR, syndrome d’Irvine-Gass ou OMD chronique (fig. 4-17). Elle peut être réséquée chirurgicalement, mais le pronostic visuel est souvent moins bon que dans les membranes épirétiniennes (MER) idiopathiques.
Un remodelage vasculaire interne s’associe souvent aux OM chroniques des OVR et OMD. D’une part des macro-anévrismes capillaires ou veineux peuvent se développer et contribuer à la pérennisation de l’OM. Il est important de savoir les identifier en angiographie (fluorescéine et/ou ICG), car s’ils sont accessibles à un traitement par laser, ils peuvent une fois traités permettre la guérison de l’OM (fig. 4-18 et 4-19). D’autre part, dans les OVR, les vaisseaux de shunts développés peuvent être à l’origine de diffusions chroniques difficiles à traiter de façon définitive (fig. 4-20).
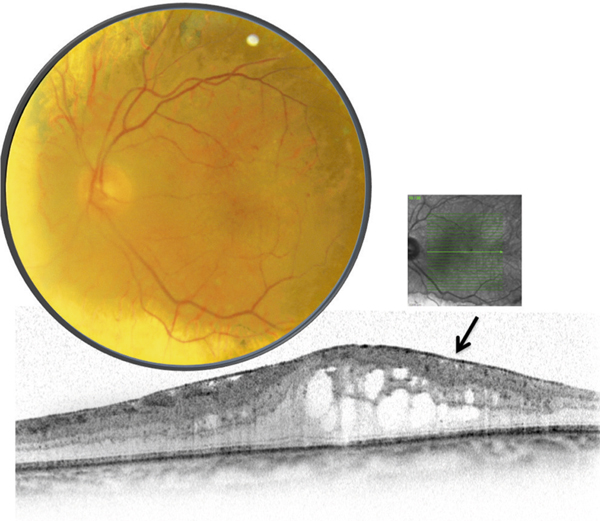
Fig. 4-17 Œdème maculaire diabétique associé à une membrane épirétinienne (flèche) chez un patient ayant eu une photocoagulation panrétinienne.
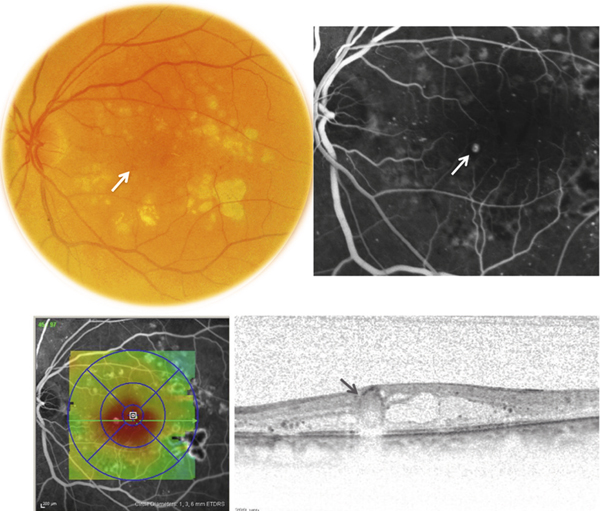
Fig. 4-18 Œdème maculaire diabétique chronique réfractaire multitraité, associé à un volumineux micro-anévrisme (flèches).
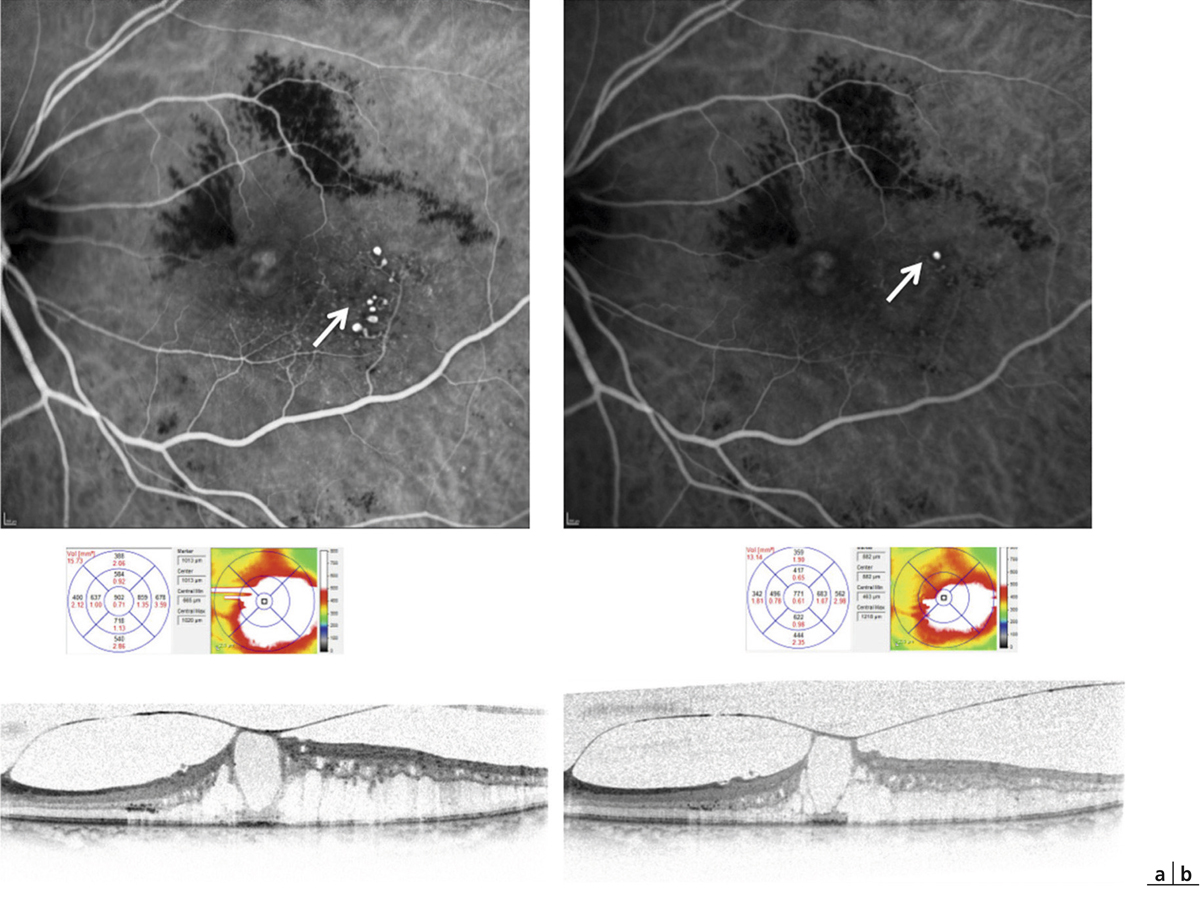
Fig. 4-19 Œdème maculaire chronique secondaire à une occlusion de branche veineuse rétinienne avec syndrome de traction vitréomaculaire.
a. Angiographie au vert d’indocyanine montrant de nombreux macro-anévrismes (flèche) accessibles au laser. b. Un mois après une première séance de laser, la majorité des macro-anévrismes ont été occlus, il ne persiste qu’un seul macro-anévrisme encore perméable. L’épaisseur maculaire a nettement diminué.
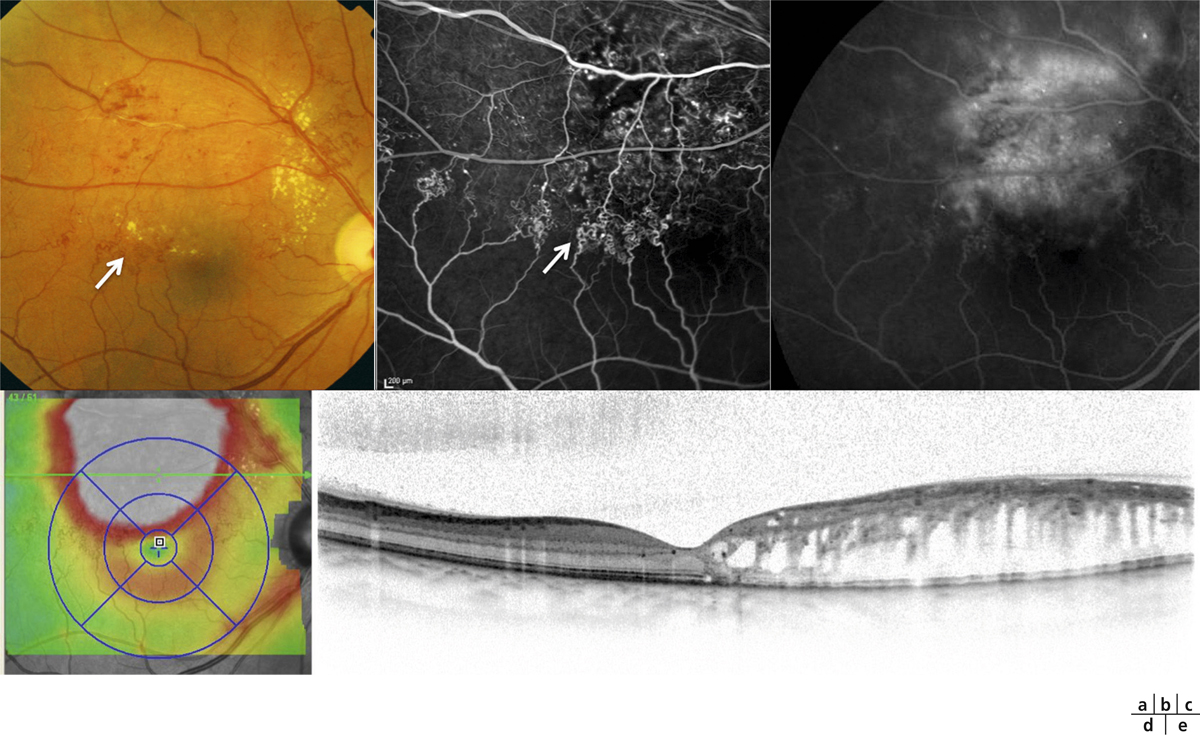
Fig. 4-20 Œdème maculaire chronique secondaire à une occlusion de branche veineuse rétinienne.
Les télangiectasies secondaires sont bien visibles sur le cliché en couleurs (a) et en angiographie à la fluorescéine (b, flèches), où elles sont source de diffusions importantes au temps tardif (c). d, e. Cartographie et OCT correspondant.
Un remaniement des couches externes est fréquemment retrouvé [12]. En OCT, la disparition de la ligne ellipsoïde (anciennement ligne inner and outer segment ou IS-OS) et de la membrane limitante externe (constituant la terminaison profonde des cellules gliales de Müller faisant synapse avec les segments internes des photorécepteurs) pourrait constituer un marqueur de dégénérescence neuronale. La désorganisation en OCT des couches rétiniennes maculaires (disorganisation of retinal inner layers [DRIL]) est également un témoin d’une altération architecturale transrétinienne secondaire à l’OMD [13] et pourrait représenter un facteur de pronostic visuel. Différentes anomalies architecturales rencontrées après résolution d’un OM chronique sont illustrées dans la figure 4-21.
Les tubulations rétiniennes, décrites notamment dans la dégénérescence maculaire liée à l’âge (DMLA) et la CRSC chronique, témoignent de remaniements atrophiques de la rétine externe soumise à un œdème intrarétinien. Elles font suite à des phénomènes exsudatifs chroniques affectant la rétine externe, mais ne constituent pas un signe d’œdème actif.
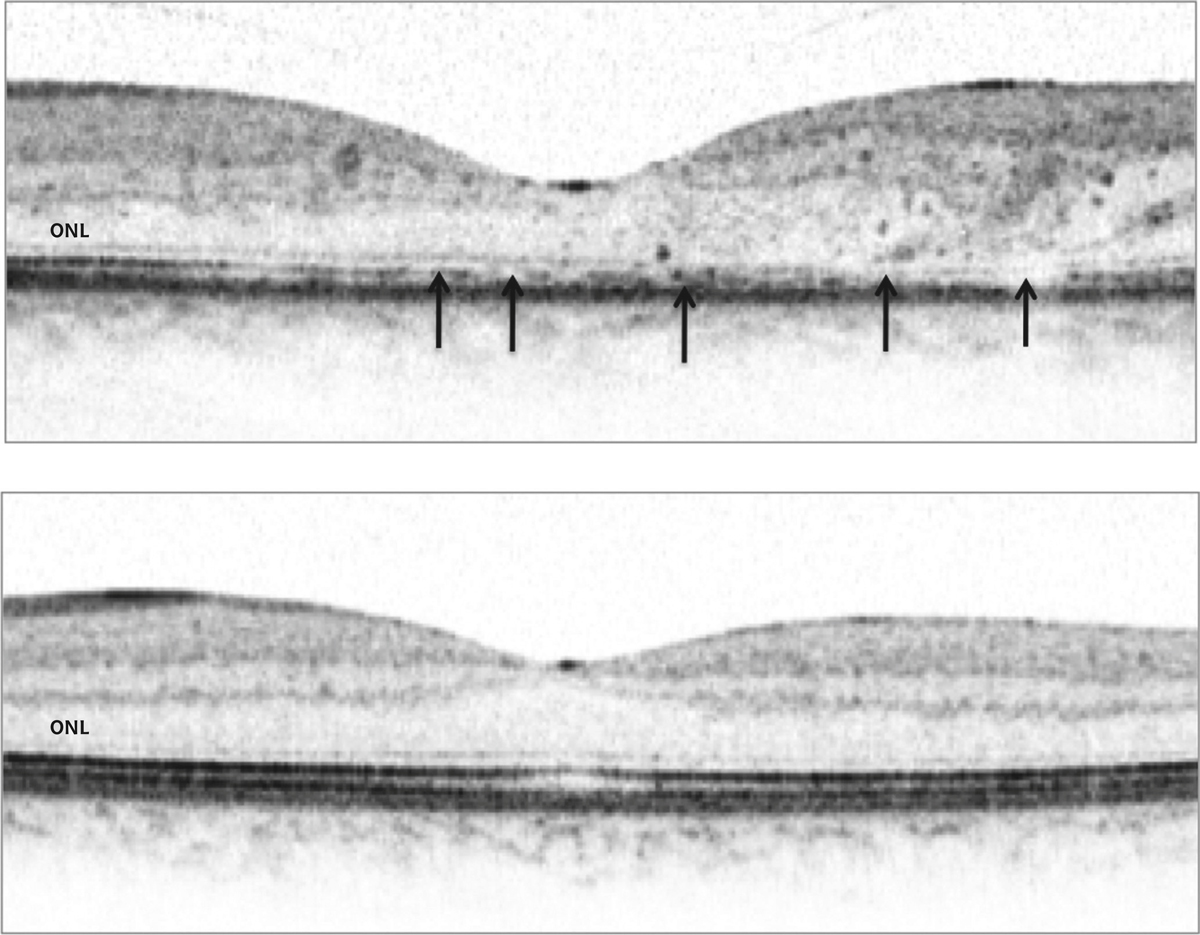
Fig. 4-21 Aspect en spectral-domain optical coherence tomography (SD-OCT) du remaniement de l’architecture maculaire après résolution d’un œdème maculaire secondaire à une occlusion veineuse rétinienne.
Haut : zones d’interruption des lignes ellipsoïdes et d’interdigitation (flèches) associées à une désorganisation des couches de la rétine interne et externe. Bas : aspect OCT d’une rétine saine.
Selon la durée de l’OM et le degré de pertes cellulaires, la fonction visuelle sera plus ou moins affectée. Dans les œdèmes inflammatoires de type Irvine-Gass, où l’OM est aigu et le plus souvent de courte durée, la récupération visuelle est bonne, car la dysfonction neurogliale n’est que transitoire et réversible, alors que dans les pathologies chroniques comme les OVR ou l’OMD, l’acuité visuelle (AV) finale est généralement diminuée. L’AV n’est que peu corrélée au degré d’épaississement maculaire [14], car elle dépend de la quantité de neurones fonctionnels au moment où est effectuée l’évaluation de la fonction visuelle [4]. La dysfonction neuronale induite par l’OM prend différents aspects :
-
l’effet de diffraction lumineuse (effet Stiles-Crawford) est la propriété des cônes de l’œil humain à présenter une sensibilité directionnelle au faisceau lumineux incident. Une lumière passant en marge de la pupille, et non parallèle à l’axe des photorécepteurs, sera donc moins efficace à stimuler la rétine qu’une lumière passant par le centre de la pupille. La présence de liquide intrarétinien modifie donc les propriétés optiques de l’œil dès les premiers stades de l’OM, avant même qu’une dégénérescence neurogliale ne soit constatée ;
-
la sensibilité aux contrastes, basée sur la capacité du système visuel à détecter des différences de luminance entre l’objet et le fond, peut également être altérée en cas d’atteinte maculaire. Elle constituerait un moyen complémentaire et sensible pour détecter une maculopathie débutante [15] ;
-
une baisse de la sensibilité rétinienne peut également être constatée à des degrés plus ou moins avancés d’OM [16], le seuil de sensibilité diminuant de façon inversement proportionnelle à l’épaisseur rétinienne ;
-
le décentrage du point de fixation, associé à une perte de sa stabilité, est également consécutif à une maculopathie, et contribue à altérer la qualité visuelle [17]. En cas d’OM post-OVR, la normalisation de l’épaisseur maculaire après injection intravitréenne de corticoïdes s’associe à un recentrage du point de fixation, ainsi qu’à une amélioration de la sensibilité rétinienne (fig. 4-22) [18] ;
-
la baisse d’AV à proprement parler est cependant lente et est souvent constatée plus tardivement que les anomalies décrites ci-dessus. Dans l’étude Early Treatment Diabetic Retinopathy Study (ETDRS), 24 % des patients avec un OMD avaient perdu plus de 3 lignes d’AV après 3 ans de suivi [19] ;
-
les métamorphopsies sont quant à elles plus rares dans l’OM simple et souvent associées à des membranes épirétiniennes développées secondairement.
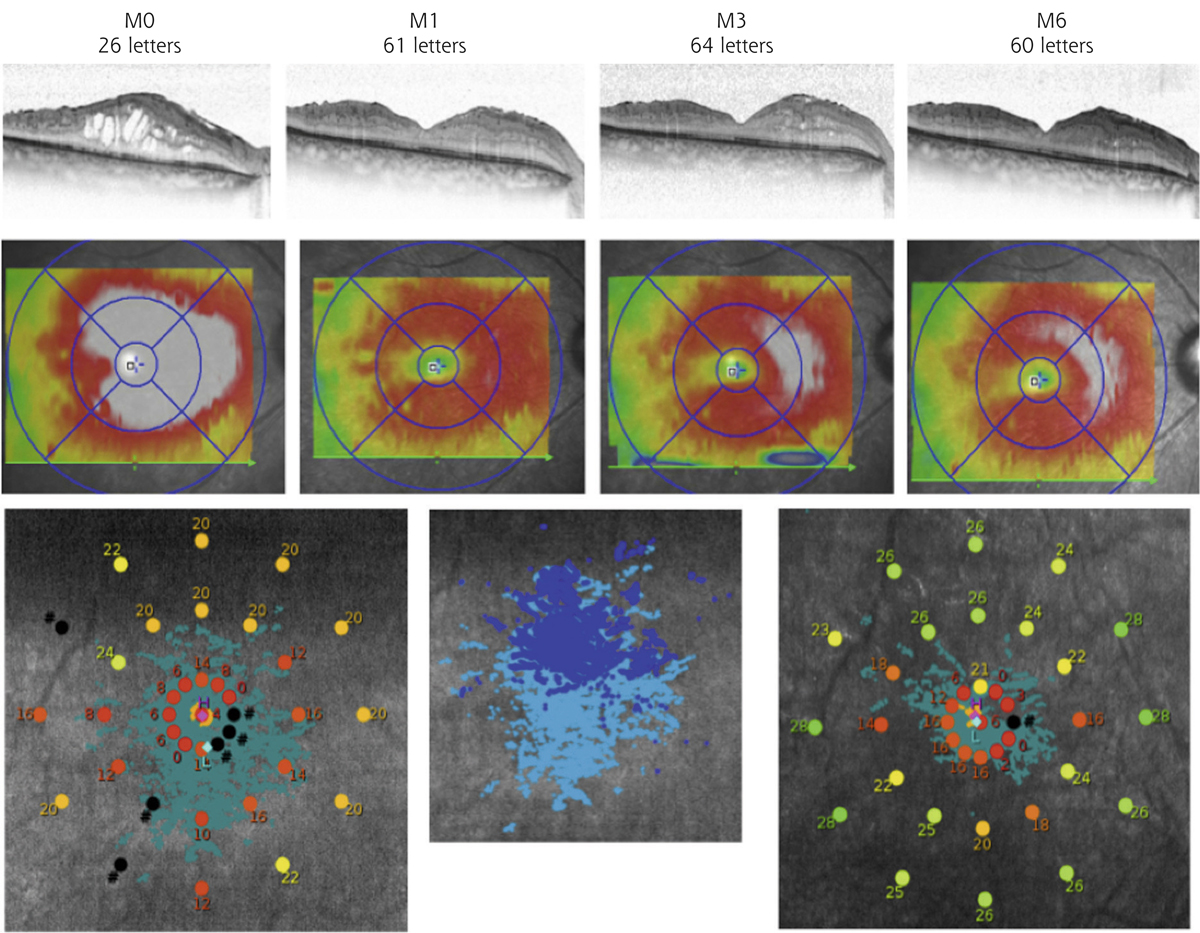
Fig. 4-22 Évolution des seuils de sensibilité et du point de fixation avant (M0) et après traitement pharmacologique d’un œdème maculaire diabétique (M1, M3, M6).
Ligne du haut : SD-OCT aux différents temps de suivi. Ligne du milieu : cartographies maculaires correspondantes. Ligne du bas : micropérimétrie avant et après traitement. Le nuage de points bleu clair correspond à la fixation avant traitement, le nuage de points bleu foncé à celui après résolution de l’OM. Noter que ce dernier est plus homogène et resserré après assèchement rétinien. La sensibilité rétinienne est également augmentée.
Les conséquences fonctionnelles et organiques de l’OM sur la rétine ne sont pas corrélées de façon linéaire. La récupération visuelle dépend du degré de perte neurogliale induite par l’OM, souvent lié à la durée d’évolution de la maladie causale. Si les traitements intravitréens permettent souvent de restituer une épaisseur maculaire normale en OCT, les modifications architecturales séquellaires de l’OM restent au premier plan pour expliquer le caractère incomplet de la récupération visuelle. Des tests de fonction visuelle, alternatifs à la simple mesure de l’acuité visuelle, semblent importants à effectuer pour évaluer l’étendue de la gêne fonctionnelle et le retentissement sur la qualité de vie du patient.
[1] Fine BS, Brucker AJ. Macular edema and cystoid macular edema. Am J Ophthalmol 1981 ; 92 : 466-81.
[2] Verbraak FD. Neuroretinal degeneration in relation to vasculopathy in diabetes. Diabetes 2014 ; 63 : 3590-2.
[3] Gass JD. A fluorescein angiographic study of macular dysfunction secondary to retinal vascular disease. IV. Diabetic retinal angiopathy. Arch Ophthalmol 1968 ; 80 : 583-91.
[4] Pelosini L, Hull CC, Boyce JF, et al. Optical coherence tomography may be used to predict visual acuity in patients with macular edema. Invest Ophthalmol Vis Sci 2011 ; 52 : 2741-8.
[5] Tsujikawa A, Sakamoto A, Ota M, et al. Serous retinal detachment associated with retinal vein occlusion. Am J Ophthalmol 2010 ; 149 : 291-301.
[6] Horii T, Murakami T, Nishijima K, et al. Relationship between fluorescein pooling and optical coherence tomographic reflectivity of cystoid spaces in diabetic macular edema. Ophthalmology 2012 ; 119 : 1047-55.
[7] Weinberger D, Fink-Cohen S, Gaton DD, et al. Non-retinovascular leakage in diabetic maculopathy. Br J Ophthalmol 1995 ; 79 : 728-31.
[8] Ota M, Nishijima K, Sakamoto A, et al. Optical coherence tomographic evaluation of foveal hard exudates in patients with diabetic maculopathy accompanying macular detachment. Ophthalmology 2010 ; 117 : 1996-2002.
[9] Gaucher D, Sebah C, Erginay A, et al. Optical coherence tomography features during the evolution of serous retinal detachment in patients with diabetic macular edema. Am J Ophthalmol 2008 ; 145 : 289-96.
[10] Jackson GR, Scott IU, Quillen DA, et al. Inner retinal visual dysfunction is a sensitive marker of non-proliferative diabetic retinopathy. Br J Ophthalmol 2012 ; 96 : 699-703.
[11] Bonnin S, Tadayoni R, Erginay A, et al. Correlation between ganglion cell layer thinning and poor visual function after resolution of diabetic macular edema. Invest Ophthalmol Vis Sci 2015 ; 56 : 978-82.
[12] Murakami T, Yoshimura N. Structural changes in individual retinal layers in diabetic macular edema. J Diabetes Res 2013 ; 2013 : 920713.
[13] Sun JK, Radwan S, Soliman AZ, et al. Neural retinal disorganization as a robust marker of visual acuity in current and resolved diabetic macular edema. Diabetes 2015 ; 64 : 2560-70.
[14] Diabetic Retinopathy Clinical Research Network, Browning DJ, Glassman AR, Aiello LP, et al. Relationship between optical coherence tomography-measured central retinal thickness and visual acuity in diabetic macular edema. Ophthalmology 2007 ; 114 : 525-36.
[15] Dorr M, Lesmes LA, Lu ZL, Bex PJ. Rapid and reliable assessment of the contrast sensitivity function on an iPad. Invest Ophthalmol Vis Sci 2013 ; 54 : 7266-73.
[16] Hatef E, Colantuoni E, Wang J, et al. The relationship between macular sensitivity and retinal thickness in eyes with diabetic macular edema. Am J Ophthalmol 2011 ; 152 : 400-5.e2.
[17] Morales MU, Saker S, Mehta RL, et al. Preferred retinal locus profile during prolonged fixation attempts. Can J Ophthalmol 2013 ; 48 : 368-74.
[18] Senturk F, Ozdemir H, Karacorlu M, et al. Microperimetric changes after intravitreal triamcinolone acetonide injection for macular edema due to central retinal vein occlusion. Retina 2010 ; 30 : 1254-61.
[19] Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Arch Ophthalmol 1985 ; 103 : 1796-806.
X. Guillonneau, F. Sennlaub
➤ Les cytokines sont des molécules de signalisation intercellulaire impliquées dans la rupture des barrières hémato-rétiniennes (BHR) et l’inflammation.
➤ Les principales cytokines impliquées dans la pathogénie des OM sont le vascular endothelial growth factor (VEGF), les interleukines (IL) 6 et 8, et le chemokine ligand 2 (CCL2) aussi connu comme monocyte chemotactic protein-1 (MPC-1).
➤ L’expression du VEGF est régulée par les conditions d’oxygénation du tissu et un grand nombre de médiateurs inflammatoires. C’est un puissant facteur d’hyperperméabilité et il a une action pro-inflammatoire.
➤ Si des taux élevés d’IL-6 et d’IL-8 sont retrouvés dans les fluides oculaires de patients atteints d’OM, leur mécanisme d’action dans la formation de l’OM n’est pas élucidé.
➤ CCL-2 pourrait induire un recrutement de monocytes dans la rétine, eux-mêmes source importante de cytokines inflammatoires.
➤ Le VEGF ne peut pas à lui seul expliquer la physiopathologie des OM et des mécanismes indépendants du VEGF prenant part à l’OM.
Les œdèmes maculaires diabétique (OMD) et les œdèmes consécutifs à une occlusion veineuse (OVR) se caractérisent par une rupture de la BHR associée à une inflammation locale. Les mécanismes moléculaires sous-tendant ces deux manifestations cliniques impliquent des molécules de signalisation intercellulaire appelées cytokines qui regroupent les familles des facteurs de croissance, les interleukines et les chimiokines. Ces cytokines sont des protéines multifonctionnelles qui peuvent être impliquées dans la prolifération, la survie ou au contraire la mort cellulaire. Elles ont également un rôle important dans la migration des cellules du système immunitaire et leur niveau d’activation. Le rôle de chacune de ces cytokines dans la perméabilité vasculaire et/ou l’inflammation rétinienne est généralement encore mal compris en raison de la variabilité clinique de ces pathologies, du grand nombre de cytokines impliquées et de la redondance de leur fonction.
Dans une situation pathologique complexe telle que la rétinopathie diabétique proliférative (RDP), mêlant ischémie, œdème, néovascularisation et perte neuronale, une large méta-analyse de 118 études a identifié 12 cytokines qui constituent des biomarqueurs de la RDP. Cela incluait : le VEGF, l’IL-6, l’IL-8 et le CCL2 [1]. Le rôle de ces cytokines dans l’apparition de l’œdème peut être abordé de façon plus pertinente chez les patients présentant un OMD en l’absence de néovascularisation ou consécutif à une occlusion veineuse.
Un nombre plus restreint d’études a quantifié les taux de cytokines présentes dans l’humeur aqueuse ou dans le vitré de patients souffrant d’un OM à l’aide de dosage enzyme-linked immunosorbent assay ou ELISA (technique quantitative basée sur l’immunodétection de la molécule cible à l’aide d’un anticorps spécifique) ou de dosage multiplex selon la technologie Luminex® (technique basée sur l’immunodosage quantitatif simultané d’un panel de cytokines à l’aide d’anticorps couplés à des microbilles fluorescentes d’une couleur spécifique pour chaque anticorps). Les principales caractéristiques de ces études sont résumées dans le tableau 4-1. Bien que ces études portent sur des cohortes de taille très modeste, les variations des taux de cytokines rapportées nous permettent de mieux appréhender la physiopathologie des œdèmes maculaires. L’ensemble de ces études indique une augmentation du facteur de croissance VEGF, des IL-6 et IL-8 et de la chimiokine CCL2 (tableaux 4-2 et 4-3) dans les OMD et les OM consécutifs à une OVR.
Tableau 4-1 Principales études sur les taux de cytokines intra-oculaires associés à un oedème maculaire.

Tableau 4-2 Taux des principales cytokines intra-oculaires lors des rétinopathies diabétiques.

Tableau 4-3 Principales cytokines intra-oculaires lors des occlusions veineuses de la rétine.
La famille du VEGF est composée de six membres dont le VEGF-A (ci-après nommé VEGF). Le VEGF est un facteur mitotique des cellules endothéliales, mais il est aussi impliqué dans la vasodilatation, la survie et la migration des cellules endothéliales [2]. Dans la rétine, il est produit par les cellules de l’épithélium pigmenté, les péricytes, les astrocytes, les cellules de Müller et les cellules endothéliales. En conditions pathologiques, les cellules du système inflammatoire, dont les monocytes/macrophages, sont une importante source de VEGF. L’expression du VEGF est régulée par les conditions d’oxygénation du tissu et un grand nombre de médiateurs inflammatoires. L’hypoxie régule à la fois la stabilité et le niveau d’expression de l’acide ribonucléique messager (ARNm) du VEGF. L’expression du VEGF peut également être augmentée par le transforming growth factor β (TGF-β), le tumor necrosis factor α (TNF-α), l’insulin-like growth factor-1 (IGF-1), de nombreuses interleukines, les produits de glycation avancés et les espèces oxygénées réactives (pour revue [3]). L’ensemble des voies de signalisation n’est pas encore compris. Cependant la voie du « signal transducer and activator of transcription factor 3 » (STAT3) occupe une place prépondérante dans cette signalisation [4].
Une augmentation significative du VEGF dans les fluides oculaires (humeur aqueuse ou vitré) a été montrée dans plusieurs études indépendantes (tableau 4-1) dans le cas d’OMD (tableau 4-2) [5–8]. Seuls Yoshimura et al. n’ont pas observé d’augmentation du taux de VEGF sur une cohorte de 92 patients atteints d’OM par rapport à un groupe contrôle de 81 patients opérés de trou maculaire [9]. Selon les études, le taux de VEGF des patients atteints de rétinopathie diabétique non proliférante (RDNP) est inférieur [7] ou supérieur [8] à celui des patients atteints de rétinopathie diabétique proliférante RDP (tableau 4-2). De même, une augmentation des concentrations de VEGF dans l’humeur aqueuse ou le vitré a été observée chez des patients atteints d’occlusion de la veine centrale de la rétine (OVCR) [9–11] ou d’occlusion d’une branche veineuse rétinienne (OBVR) [6, 10–12]. Cependant Funk et al. et Yoshimura et al. n’ont pas mis en évidence d’augmentation significative du VEGF chez les patients atteints d’OBVR (tableau 4-3) [9, 13].
L’augmentation de la perméabilité vasculaire est un des points clés des rétinopathies et participe à la formation de l’œdème. Cette augmentation de perméabilité est observée chez les patients diabétiques et dans les OVR ainsi que dans les modèles animaux de ces pathologies. Le VEGF induit cette perméabilité par un mécanisme biphasique. Dans un premier temps, le VEGF augmente la perméabilité transcellulaire en augmentant le transport par transcytose de vésicules de cavéoline [14]. À plus long terme, le VEGF augmente la perméabilité vasculaire en altérant l’expression des molécules impliquées dans les jonctions serrées [14].
Les taux de VEGF sont généralement corrélés à la gravité de l’OMD [5, 7]. Cependant Lee et al. et Roh et al. n’ont pas pu mettre en évidence cette corrélation dans leur étude [6, 15]. Dans le cas des OVR, les taux de VEGF sont corrélés principalement à la surface d’ischémie liée à l’occlusion. Les taux de VEGF sont retrouvés corrélés [5, 16] ou non [10] à l’épaisseur rétinienne maculaire. Enfin, le taux de VEGF n’est pas nécessairement lié aux taux des autres cytokines inflammatoires de la rétine [9] suggérant un rôle autonome de celui-ci dans le déclenchement de l’œdème.
L’injection d’anti-VEGF conduit à un gain d’acuité visuelle et à une diminution de l’épaisseur maculaire [17]. L’injection de bévacizumab chez des patients atteints d’OM associé à des RDP ou RDNP induit systématiquement une diminution du taux de VEGF [13, 15, 18, 19]. Cependant, seuls Sohn et al. ont pu mettre en évidence une liaison statistique entre la normalisation du VEGF et la disparition de l’œdème maculaire diabétique et une amélioration de l’acuité visuelle [19]. Deux autres études sur des cohortes de petite taille, dans lesquelles a été mesuré le taux de VEGF après injection de bévacizumab, n’ont pas pu mettre en évidence de lien entre le taux de VEGF après injection, la disparition de l’œdème maculaire diabétique et l’amélioration de l’acuité visuelle [15, 18]. Ces injections ne modifient pas le taux des cytokines IL-6, IL-8 et CCL2 chez les patients naïfs après la première injection [15, 18]. En revanche, une diminution du taux de IL-6 et de CCL2 a été observée chez les patients ayant déjà reçu plusieurs injections de bévacizumab [15].
L’injection de bévacizumab dans les cas d’OBVR (ayant un taux de VEGF comparable aux contrôles) et d’OVCR (taux de VEGF supérieur aux contrôles) permet de diminuer les taux de VEGF en dessous du taux détectable et d’améliorer ainsi l’acuité visuelle et l’épaisseur rétinienne [13]. Kaneda et al. dans une étude portant sur 38 patients atteints d’OBVR traités par bévacizumab n’ont pas pu mettre en évidence de corrélation entre les taux d’IL-6, d’IL-8 et de MCP-1 avant injection et les résultats après traitement anti-VEGF [20].
Sohn et al. ont montré une diminution des taux de VEGF, d’IL-6 et de CCL2 après injection de triamcinolone chez des patients atteints d’OMD, associée à une diminution de l’œdème [19]. Park et al. ont montré une diminution du taux de VEGF chez les patients répondant aux injections de triamcinolone [21].
En conclusion, si les études sur de larges cohortes démontrent un bénéfice incontestable des thérapies anti-VEGF, il est difficile d’établir un lien direct entre :
-
le taux de VEGF et la pathologie causale ;
-
la diminution effective du taux de VEGF et le succès du traitement anti-VEGF.
Le rôle d’IL-6 dans la rétinopathie diabétique est encore largement méconnu en partie à cause des multiples rôles de cette cytokine. IL-6 n’est que très faiblement exprimée par les cellules microgliales dans la rétine non pathologique [22]. IL-6 a été initialement caractérisée comme un inducteur de la production d’anticorps par les lymphocytes B. Son activité sur le système immunitaire est en fait bien plus complexe et inclut, par exemple, la différenciation des lymphocytes Th, l’induction de la fièvre et la production d’autres interleukines. Dans le système nerveux, son rôle est également complexe puisque IL-6 peut apparaître soit comme neuroprotecteur, soit comme acteur de la neuro-inflammation [23].
La concentration d’IL-6 a été trouvée significativement élevée dans les fluides oculaires de patients atteints d’OMD (tableau 4-2) [5, 6, 9], d’OVCR [9–11] et d’OBVR (tableau 4-3) [9–12, 20]. Seule l’étude de Lee et al. ne note pas d’augmentation d’IL-6 chez des patients atteints d’occlusion de branche veineuse [6]. Le mécanisme d’action d’IL-6 dans la formation de l’œdème n’est pas encore connu. L’injection d’IL-6 dans un modèle murin induit une leucostase importante qui est une des caractéristiques de la rétinopathie diabétique. L’expression d’IL-6 est nécessaire à l’effet biologique de l’angiotensine 2 [22]. Une corrélation positive entre le taux d’IL-6 et l’épaisseur maculaire a été montrée par Feng et al. et Noma et al. [10, 12]. Bien qu’absente en condition normale, IL-6 peut être exprimée en condition pathologique par les cellules de Müller, les cellules endothéliales, les cellules épithéliales pigmentées et toutes les cellules de la lignée myéloïde. La source précise d’IL-6 durant l’OMD n’est pas connue, il paraît vraisemblable que les cellules de Müller et les cellules inflammatoires soient les principales sources de cette cytokine.
Malgré une évidente corrélation entre la pathologie et le taux d’IL-6 présent dans les fluides oculaires [24], il reste difficile d’établir un bénéfice thérapeutique à la diminution d’IL-6 dans les OM. L’injection de bévacizumab ne réduit pas le taux d’IL-6 [13, 15, 19] malgré le succès évident de cette thérapie. La réduction du taux d’IL-6 ne constitue donc pas un facteur du succès de ces thérapies [25]. Dans le cas des traitements anti-inflammatoires, les résultats restent également non concluants alors que ces traitements diminuent efficacement le taux d’IL-6 [19, 21]. Chez les patients atteints d’OMD, l’injection intravitréenne de triamcinolone réduit le taux d’IL-6 chez tous les patients, qu’ils soient répondeurs ou non au traitement, mais une persistance de l’œdème est observée parmi les patients conservant un taux élevé de VEGF malgré le traitement [21]. Seul Roh et al. retrouvent une corrélation négative entre le taux de VEGF et le taux d’IL-6, une semaine après injection d’anti-VEGF chez des patients ne présentant pas d’amélioration significative de l’œdème après l’injection [15].
En conclusion, bien qu’un taux élevé d’IL-6 soit observé dans l’immense majorité des études, il est difficile de dire, d’après les données actuelles, si son inhibition présente un intérêt thérapeutique potentiel. Sur la base de l’activité biologique de l’IL-6 dans la perméabilité vasculaire et des études ci-dessus, un antagoniste de l’IL-6 est en phase d’essai préclinique pour une potentielle utilisation dans les OMD [26]. Le succès ou non de cette nouvelle voie thérapeutique donnera une indication sur le rôle d’IL-6 dans cette pathologie.
IL-8 est une cytokine à la fois pro-inflammatoire et pro-angiogénique. Elle permet d’induire la transmigration des polynucléaires neutrophiles à travers la barrière hémato-encéphalique [27]. Dans le cerveau, les polynucléaires neutrophiles participent à l’augmentation de la perméabilité vasculaire et à la formation de l’œdème en condition pathologique [28, 29]. Il-8 est également directement impliquée dans l’angiogenèse et la perméabilité vasculaire par un mécanisme non lié à l’hypoxie [30].
Il-8 est exprimée par les cellules du système immunitaire mais également par les cellules endothéliales et les cellules de l’épithélium pigmenté [31]. L’expression du VEGF et celle de l’IL-8 sont interdépendantes : IL-8 augmente l’expression du VEGF des cellules endothéliales [30], mais il a également été montré sur des cellules microvasculaires que le VEGF induisait l’expression d’IL-8 et la transmigration des neutrophiles [32].
La concentration d’IL-8 est trouvée significativement élevée dans les fluides oculaires de patients atteints d’OMD (tableau 4-2) [5, 6, 9], d’OVCR [9–11] et d’OBVR (tableau 4-3) [6, 9, 10, 20].
Son rôle dans la survenue de l’œdème rétinien n’est pas encore totalement compris. Song et al. ont trouvé une corrélation entre le niveau d’activation des neutrophiles et le degré de sévérité de la rétinopathie [33], tandis que Joussen et al. ont montré que les neutrophiles étaient responsables d’une augmentation de la perméabilité vasculaire et de la perte capillaire dans un modèle de rongeur diabétique [34]. Deux études ont montré, à la fois dans les OMD et les OBVR, que le taux d’IL-8 était positivement corrélé à la sévérité de l’OM [6, 20].
Bien que VEGF et IL-8 puissent se réguler de façon croisée [30, 32], plusieurs études ont montré une indépendance statistique entre l’augmentation de VEGF et d’IL-8 suggérant un rôle direct d’IL-8 dans la survenue de l’œdème.
L’injection de bévacizumab chez des patients atteints d’OM associé à des RDP ou RDNP ne modifie pas le taux d’IL-8 après la première injection [15, 18] que ce soit chez les patients naïfs ou ceux ayant déjà reçu plusieurs injections de bévacizumab [15]. Cependant un polymorphisme du promoteur d’IL-8 (–251A/T), responsable de l’augmentation de la sécrétion de cette cytokine, est statistiquement associé à l’absence de réponse au bévacizumab en cas de DMLA [35]. Par ailleurs, Jeon et al. ont montré, sur une cohorte de patients non-répondeurs aux anti-VEGFs, que le taux d’IL-8 (après traitement par bévacizumab) était corrélé au taux de succès des injections de triamcinolone, suggérant à nouveau qu’IL-8 est impliquée indépendamment du VEGF dans la formation de l’OM.
En conclusion, IL-8 est une cytokine particulièrement attractive pour expliquer la survenue de l’OM. En effet, IL-8 est systématiquement augmentée dans les OM indépendamment du taux de VEGF. Les patients non-répondeurs aux anti-VEGFs sont caractérisés par un taux élevé d’IL-8. La réduction du taux d’IL-8 chez ces patients est associée à une diminution de l’œdème [25, 35]. Cependant, il n’existe pour l’instant aucune solution thérapeutique ciblant spécifiquement IL-8 dans les OM. Un anticorps humanisé anti-IL-8 est disponible depuis le milieu des années 2000. Cet anticorps n’est pour l’instant pas approuvé pour une application thérapeutique en dépit de plusieurs essais cliniques.
CCL2 (aussi connu comme MCP-1) est impliqué dans le recrutement des monocytes depuis la moelle osseuse dans les tissus lésés. Ces monocytes peuvent altérer les barrières hémato-céphaliques [36]. CCL2 est faiblement exprimé dans la rétine non pathologique et peut être produit par les cellules de Müller ou les phagocytes mononucléés (microglie, macrophages et monocytes) en conditions pathologiques [37, 38]. En cas de rétinopathie diabétique, Rangasamy et al. ont suggéré que CCL2 pouvait être produit par les cellules endothéliales [39].
La concentration de CCL2 est significativement élevée dans les fluides oculaires de patients atteints d’OMD (tableau 4-2) [5, 6, 9], d’OVCR [9–11] et d’OBVR (tableau 4-3) [9, 11, 12]. Seuls Lee et al. n’ont pas mis en évidence de variation de CCL2 chez les patients atteints d’OBVR [6]. Le taux de CCL2 n’est généralement pas positivement corrélé à la sévérité de l’OVR [6, 20, 40], alors que l’étude de Kaneda et al. portant sur 38 patients atteints d’OBVR indique quant à elle une corrélation positive [40].
Cette augmentation de CCL2 pourrait induire un recrutement important de phagocytes mononucléés dans la rétine. À l’appui de cette hypothèse, une augmentation du nombre de phagocytes mononucléés et une activation gliale ont été notées à la fois dans les modèles de rétinopathie diabétique et chez les patients atteints de rétinopathie diabétique précoce [39, 41–43]. CCL2 pourrait également directement modifier la barrière hémato-rétinienne [39, 44] ; cependant les travaux de Rangasamy et al. suggèrent plutôt un rôle indirect de CCL2 qui agirait en recrutant des monocytes, source importante de cytokines inflammatoires (IL-1, IL-6 et TNF-α) modifiant la perméabilité vasculaire [39]. Une corrélation positive entre le taux de CCL2 et la sévérité de l’OM a été montrée en cas d’OM [5] consécutif à une OBVR [40]. Même si plusieurs études n’ont pas pu mettre en évidence ce lien statistique [6, 10, 16, 20], il semble donc que CCL2 joue un rôle important dans la rupture de la barrière hémato-rétinienne.
Un taux élevé de CCL2 est retrouvé dans la grande majorité des études, et ce indépendamment du taux de VEGF [15]. Dans le cas d’OMD, l’injection de bévacizumab ne modifie pas le taux de CCL2 [15, 18], mais une corrélation négative entre le taux de VEGF et de CCL2 a été notée après plusieurs injections de bévacizumab [15]. Dans le cas des OVR, les traitements anti-VEGF peuvent affecter le taux de CCL2 [13] et les injections de triamcinolone diminuent le taux de CCL2, mais cette diminution n’est pas statistiquement liée à la réduction de l’épaisseur rétinienne [40].
En conclusion, tout comme IL-6, une augmentation de CCL2 est observée dans l’immense majorité des études. Il existe un véritable rationnel scientifique indiquant que les cellules de la lignée myéloïde participent à la pathogénie des OM, cependant les études menées jusqu’à présent n’ont pas permis d’établir un lien direct entre CCL2 et œdème. Des études de plus grande taille permettront certainement dans le futur de mieux comprendre le rôle et l’intérêt thérapeutiques de limiter la bio-activité de CCL2 dans ces pathologies.
Enfin, certaines cytokines sont trouvées dans l’humeur aqueuse ou le vitré de patients atteints d’OM, sans que ces résultats ne soient reproduits dans un nombre significatif d’études indépendantes. Ces cytokines ont des rôles connus dans l’inflammation et la perméabilité vasculaire et pourraient être impliquées dans le développement de l’OM. C’est le cas du platelet-derived growth factor (PDGF) AA et AB dont la concentration est augmentée en cas d’OM, alors qu’une diminution du taux de pigment epithelium-derived growth factor (PEDGF) BB est observée [6, 7]. Le pigment epithelium derived factor (PEDF) est retrouvé diminué chez les patients atteints d’OM [5, 8] ou d’OVR [12]. Cette diminution du PEDF est corrélée avec la surface de rétine non perfusée et avec l’épaisseur maculaire [12]. Enfin Feng et al. ont retrouvé une augmentation d’IL-1β, de TGF-β et de fibroblast growth factor 2 (FGF-2) dans l’humeur aqueuse de patients atteints d’OVCR et d’OBVR (tableau 4-3) [10].
Les OM se caractérisent par une modification importante des taux de cytokines dans les fluides oculaires, qui reflètent une situation pathologique mêlant perméabilité vasculaire et inflammation. Les augmentations de VEGF, d’IL-6, IL-8 et de CCL2 apparaissent comme les meilleurs biomarqueurs de ces OM (tableaux 4-2 et 4-3). L’apparition des anti-VEGFs a révolutionné la prise en charge de ces OM avec une efficacité thérapeutique prouvée [17]. Cependant l’analyse des fluides oculaires de patients atteints d’OM démontre une grande disparité des taux de VEFG chez ces patients, et certaines études n’ont pu démontrer d’association entre taux de VEGF et importance de l’OM [9, 18]. Cette grande variabilité indique que le VEGF ne peut, à lui seul, expliquer la physiopathologie des OM et que des mécanismes indépendants du VEGF prennent part à l’OM.
Les traitements anti-VEGF ne suffisent pas à réduire la part inflammatoire de ces pathologies même lorsqu’ils réduisent l’OM. En laissant perdurer une situation d’inflammation, les traitements anti-VEGF laissent un risque potentiel de récidive ou de complication (neuro-inflammation et/ou atteinte de l’unité neurovasculaire). À l’inverse, les traitements à base d’anti-inflammatoire tels que la triamcinolone inhibent efficacement la production de cytokines inflammatoires, mais échouent à réduire complètement les taux de VEGF.
En conclusion, si les anti-VEGFs ont révolutionné la prise en charge des OM, le traitement des patients atteints d’OM pourrait être amélioré en prenant mieux en compte la partie inflammatoire de leur pathologie. Dans le futur, une thérapie personnalisée adaptée au profil de cytokines de chaque patient pourrait être envisagée, mais dans l’immédiat, l’utilisation de molécules ciblant spécifiquement IL-6, IL-8 et CCL2 en plus de la thérapie anti-VEGF serait certainement bénéfique à l’ensemble des patients. Ces molécules étant principalement produites par les leucocytes recrutés, ceux-ci semblent donc être la cible thérapeutique la plus intéressante. Un gain thérapeutique peut être ainsi espéré, soit en bloquant le recrutement des leucocytes (IL-8, CCL2), soit en bloquant leur activation et donc la production de cytokines pro-inflammatoires.
[1] McAuley AK, Sanfilippo PG, Hewitt AW, et al. Vitreous biomarkers in diabetic retinopathy : a systematic review and meta-analysis. J Diabetes Complications 2014 ; 28 : 419-25.
[2] Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol 2001 ; 280 : C1358-66.
[3] Caldwell RB, Bartoli M, Behzadian MA, et al. Vascular endothelial growth factor and diabetic retinopathy : pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev 2003 ; 19 : 442-55.
[4] Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002 ; 21 : 2000-8.
[5] Funatsu H, Noma H, Mimura T, et al. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 2009 ; 116 : 73-9.
[6] Lee WJ, Kang MH, Seong M, Cho HY. Comparison of aqueous concentrations of angiogenic and inflammatory cytokines in diabetic macular oedema and macular oedema due to branch retinal vein occlusion. Br J Ophthalmol 2012 ; 96 : 1426-30.
[7] Praidou A, Papakonstantinou E, Androudi S, et al. Vitreous and serum levels of vascular endothelial growth factor and platelet-derived growth factor and their correlation in patients with non-proliferative diabetic retinopathy and clinically significant macula oedema. Acta Ophthalmol 2011 ; 89 : 248-54.
[8] Patel JI, Tombran-Tink J, Hykin PG, et al. Vitreous and aqueous concentrations of proangiogenic, antiangiogenic factors and other cytokines in diabetic retinopathy patients with macular edema : implications for structural differences in macular profiles. Exp Eye Res 2006 ; 82 : 798-806.
[9] Yoshimura T, Sonoda KH, Sugahara M, et al. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS One 2009 ; 4 : e8158.
[10] Feng J, Zhao T, Zhang Y, et al. Differences in aqueous concentrations of cytokines in macular edema secondary to branch and central retinal vein occlusion. PLoS One 2013 ; 8 : e68149.
[11] Koss MJ, Pfister M, Rothweiler F, et al. Comparison of cytokine levels from undiluted vitreous of untreated patients with retinal vein occlusion. Acta Ophthalmol 2012 ; 90 : e98-e103.
[12] Noma H, Mimura T, Eguchi S. Association of inflammatory factors with macular edema in branch retinal vein occlusion. JAMA Ophthalmol 2013 ; 131 : 160-5.
[13] Funk M, Kriechbaum K, Prager F, et al. Intraocular concentrations of growth factors and cytokines in retinal vein occlusion and the effect of therapy with bevacizumab. Invest Ophthalmol Vis Sci 2008 ; 50 : 1025-32.
[14] Nag S, Kapadia A, Stewart DJ. Review : molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol 2011 ; 37 : 3-23.
[15] Roh MI, Kim HS, Song JH, et al. Effect of intravitreal bevacizumab injection on aqueous humor cytokine levels in clinically significant macular edema. Ophthalmology 2009 ; 116 : 80-6.
[16] Pfister M, Rothweiler F, Michaelis M, et al. Correlation of inflammatory and proangiogenic cytokines from undiluted vitreous samples with spectral domain OCT scans, in untreated branch retinal vein occlusion. Clin Ophthalmol 2013 ; 7 : 1061-7.
[17] Diabetic Retinopathy Clinical Research Network, Wells JA, Glassman AR, Ayala AR, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl J Med 2015 ; 372 : 1193-203.
[18] Funk M, Schmidinger G, Maar N, et al. Angiogenic and inflammatory markers in the intraocular fluid of eyes with diabetic macular edema and influence of therapy with bevacizumab. Retina 2010 ; 30 : 1412-9.
[19] Sohn HJ, Han DH, Lee DY, Nam DH. Changes in aqueous cytokines after intravitreal triamcinolone versus bevacizumab for macular oedema in branch retinal vein occlusion. Acta Ophthalmol 2013 ; 92 : 217-24.
[20] Kaneda S, Miyazaki D, Sasaki SI, et al. Multivariate analyses of inflammatory cytokines in eyes with branch retinal vein occlusion : relationships to bevacizumab treatment. Invest Ophthalmol Vis Sci 2011 ; 52 : 2982-8.
[21] Park SP, Ahn JK. Changes of aqueous vascular endothelial growth factor and interleukin-6 after intravitreal triamcinolone for branch retinal vein occlusion. Clin Experiment Ophthalmol 2008 ; 36 : 831-5.
[22] Rojas M, Zhang W, Lee DL, et al. Role of IL-6 in angiotensin II-induced retinal vascular inflammation. Invest Ophthalmol Vis Sci 2010 ; 51 : 1709-18.
[23] Suzuki S, Tanaka K, Suzuki N. Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J Cereb Blood Flow Metab 2009 ; 29 : 464-79.
[24] Funatsu H, Yamashita H, Ikeda T, et al. Vitreous levels of interleukin-6 and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2003 ; 110 : 1690-6.
[25] Jeon S, Lee WK. Effect of intravitreal triamcinolone in diabetic macular edema unresponsive to intravitreal bevacizumab. Retina 2014 ; 34 : 1606-11.
[26] Schmidt M, Matsumoto Y, Tisdale A, et al., Eleven Biotherapeutics Cambridge, MA. Optimized IL-6 blockade for the treatment of diabetic macular edema. Invest Ophthalmol Vis Sci 2015 ; 55 : 1062.
[27] Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology : a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 2010 ; 30 : 459-73.
[28] Kenne E, Erlandsson A, Lindbom L, et al. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation 2012 ; 9 : 17.
[29] Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke : role of inflammatory cells. J Leukoc Biol 2010 ; 87 : 779-89.
[30] Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem 2009 ; 284 : 6038-42.
[31] Ghasemi H, Ghazanfari T, Yaraee R, et al. Roles of IL-8 in ocular inflammations : a review. Ocul Immunol Inflamm 2011 ; 19 : 401-12.
[32] Lee TH, Avraham H, Lee SH, Avraham S. Vascular endothelial growth factor modulates neutrophil transendothelial migration via up-regulation of interleukin-8 in human brain microvascular endothelial cells. J Biol Chem 2002 ; 277 : 10445-51.
[33] Song H, Wang L, Hui Y. Expression of CD18 on the neutrophils of patients with diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol 2007 ; 245 : 24-31.
[34] Joussen AM, Poulaki V, Mitsiades N, et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J 2003 ; 17 : 76-8.
[35] Hautamaki A, Kivioja J, Vavuli S, et al. Interleukin 8 promoter polymorphism predicts the initial response to bevacizumab treatment for exudative age-related macular degeneration. Retina 2013 ; 33 : 1815-27.
[36] Rodrigues SF, Granger DN. Blood cells and endothelial barrier function. Tissue Barriers 2015 ; 3(1-2) : e978720.
[37] Rutar M, Natoli R, Valter K, Provis JM. Early focal expression of the chemokine Ccl2 by Muller cells during exposure to damage-inducing bright continuous light. Invest Ophthalmol Vis Sci 2011 ; 52 : 2379-88.
[38] Raoul W, Auvynet C, Camelo S, et al. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation 2010 ; 7 : 87.
[39] Rangasamy S, McGuire PG, Franco Nitta C, et al. Chemokine mediated monocyte trafficking into the retina : role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One 2014 ; 9 : e108508.
[40] Kunikata H, Shimura M, Nakazawa T, et al. Chemokines in aqueous humour before and after intravitreal triamcinolone acetonide in eyes with macular oedema associated with branch retinal vein occlusion. Acta Ophthalmologica 2010 ; 90 : 162-7.
[41] Zeng HY, Green WR, Tso MO. Microglial activation in human diabetic retinopathy. Arch Ophthalmol 2008 ; 126 : 227-32.
[42] Kermorvant-Duchemin E, Pinel AC, Lavalette S, et al. Neonatal hyperglycemia inhibits angiogenesis and induces inflammation and neuronal degeneration in the retina. PLoS One 2013 ; 8 : e79545.
[43] Gaucher D, Chiappore JA, Paques M, et al. Microglial changes occur without neural cell death in diabetic retinopathy. Vision Res 2007 ; 47 : 612-23.
[44] Stamatovic SM, Shakui P, Keep RF, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab 2005 ; 25 : 593-606.
M.D. de Smet, P.-H. Gabrielle
➤ Les cellules microgliales de la rétine sont des cellules appartenant au système immunitaire ayant un rôle d’immunomodulation. En cas de stress rétinien, elles vont s’activer et migrer au niveau de l’interface épithélium pigmentaire de la rétine (EPR)–choroïde par une voie transcellulaire au niveau de l’EPR, afin de réguler la réaction immunitaire et en particulier les manifestations inflammatoires.
➤ En cas de stress chronique ou d’aggravation, cette voie transcellulaire dysfonctionne et provoque l’accumulation de cellules microgliales dans l’espace sous-rétinien. Cette accumulation de cellules activées contribue à l’entretien et à l’aggravation de l’inflammation par la sécrétion de cytokines pro-inflammatoires et cytotoxiques participant à la rupture des BHR (interne/externe) et à la dérégulation du milieu intracellulaire des cellules gliales de Müller dans l’OM.
➤ De manière générale, la notion de « para-inflammation » est l’ensemble des mécanismes immunologiques d’adaptation à un stress tissulaire modéré dont le but est de maintenir le fonctionnement tissulaire. Si ce stress persiste ou augmente, ceux-ci ne sont plus adéquats et entraînent un dysfonctionnement tissulaire.
➤ Dans le cadre d’un processus inflammatoire débutant retrouvé dans les pathologies de l’OM, l’activation et la migration transépitheliale microgliale de l’EPR font partie de ces mécanismes d’adaptation et leur altération contribue à l’inflammation et à l’apparition ou la persistance de l’OM.
Les neurones, les cellules gliales – englobant astrocytes, cellules gliales de Müller (CGM) et microglie –, ainsi que les vaisseaux sanguins sont organisés en une unité neurovasculaire (fig. 4-23). Cette organisation est caractérisée par un contact physique intime et une intégration fonctionnelle qui facilite l’adaptation aux variations des conditions du milieu soit physiologiques soit pathologiques. Elle coordonne les besoins métaboliques, l’activité synaptique, l’apport en sang ainsi que l’évacuation des déchets cellulaires comme le glutamate, le monoxyde d’azote (nitric oxide ou NO), l’adénosine et les métabolites de l’acide arachidonique [1, 2].
L’OM est considéré actuellement comme principalement dû à une altération de la BHR interne et/ou externe, mais des récents travaux, en particulier dans la rétinopathie diabétique, suggèrent qu’une altération de l’unité neurovasculaire serait également en cause [3]. Il a également été établi qu’il existe un déséquilibre des mécanismes de régulation de l’immunité et de l’inflammation qui participerait à la pathogénie de l’œdème maculaire et à la perte de photorécepteurs [4]. La réponse immunitaire est différente selon les conditions physiopathologiques. Ainsi lors de stress modéré, des mécanismes immunologiques vont permettre de maintenir l’équilibre homéostasique et le fonctionnement tissulaire. Mais cet état n’est pas stable et si le stress devient chronique, ces mécanismes peuvent devenir mal adaptés et causer une pathologie. Ces mécanismes immunologiques d’adaptation sont regroupés sous le terme de « para-inflammation » [4].
Dans l’unité neurovasculaire, les cellules microgliales ont un rôle de surveillance des activités cellulaire et synaptique de la rétine et d’évacuation des cellules mortes. Ces cellules jouent également un rôle prépondérant dans l’immunité cellulaire rétinienne, en particulier d’immunomodulation.
Dans ce sous-chapitre, nous allons développer la fonction d’immunomodulation de l’inflammation des cellules microgliales et l’implication de son dysfonctionnement dans l’apparition de l’OM. Nous évoquerons ensuite la cascade de réactions conduisant à l’OM.
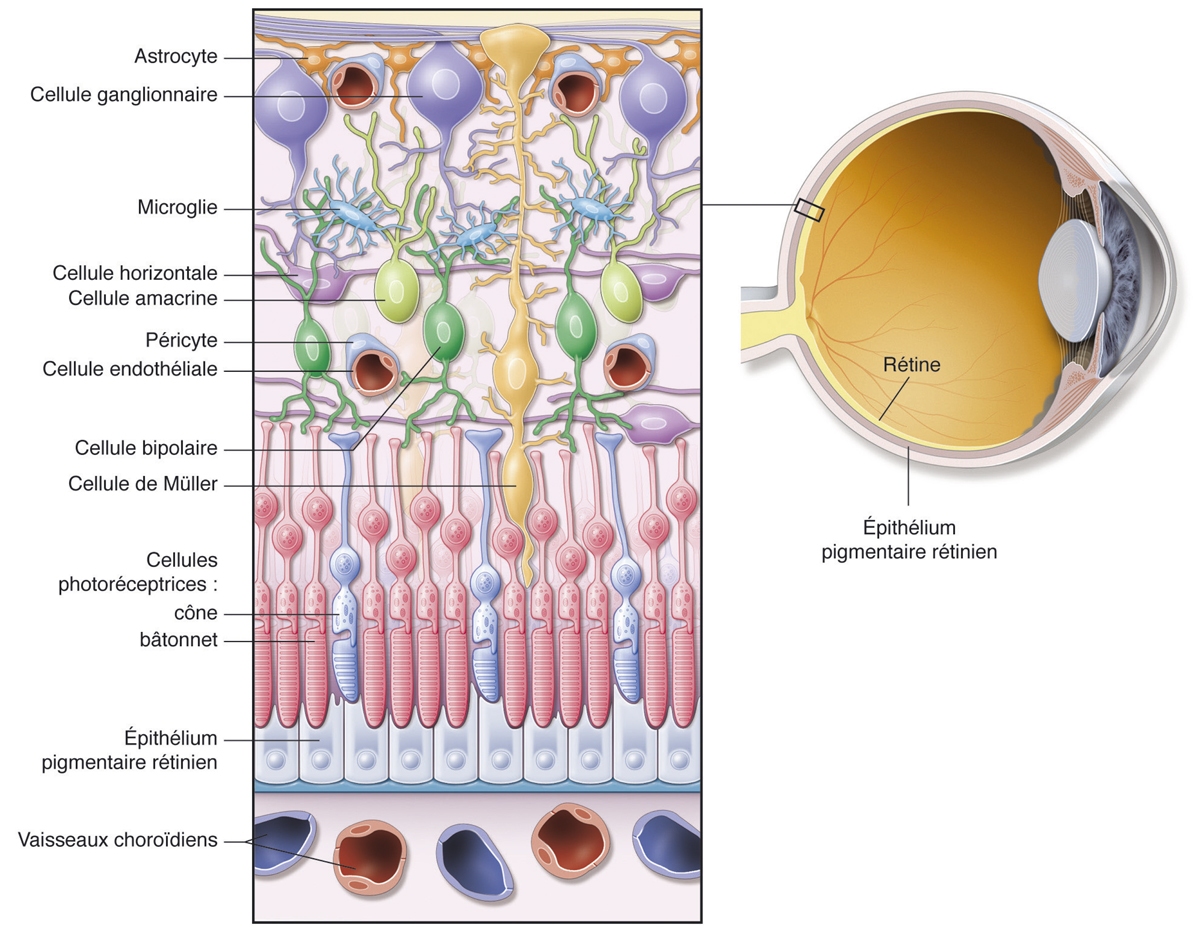
Fig. 4-23 Unité neurovasculaire.
Les neurones, la glie et les cellules vasculaires forment l’unité neurovasculaire de la rétine. Les nombreuses connexions physiologiques et anatomiques entre ces cellules permettent la vision.
Les cellules microgliales sont des cellules résidentes du système immunitaire de la rétine présentes au niveau des couches plexiformes externe et interne. Elles ont un rôle essentiel dans la défense immunitaire de la rétine [5]. Leur profil immunologique d’immunomodulation est surtout dirigé vers l’immunosuppression [6, 7], mais elles peuvent être à l’origine d’une réponse inflammatoire en cas de stress persistant [8]. Avec les macrophages périvasculaires, les cellules microgliales forment les deux types de cellules rétiniennes dérivées des monocytes et sont des cellules présentatrices d’antigènes impliquées dans la modulation de la réponse immunitaire [8]. À l’état physiologique ou en cas d’inflammation/traumatisme rétinien, des cellules précurseurs monocytaires dérivées de la moelle osseuse peuvent les renouveler [9, 10].
À l’état de repos, les cellules microgliales sont situées dans la partie interne de la rétine et ont une forme dendritique qui leur permet de sonder en permanence leur environnement grâce à de multiples prolongements cytoplasmiques (fig. 4-24 et 4-25). Elles possèdent une grande plasticité de forme en fonction de leur degré d’activation, ce qui leur permet de répondre rapidement à des situations de stress ou d’altération cellulaire, minimisant ainsi tout déséquilibre immunologique tissulaire [4]. Lorsqu’elles sont activées, elles prennent une forme amiboïde, migrent et s’accumulent dans l’espace sous-rétinien, comme, par exemple, dans la rétinopathie diabétique, l’inflammation, les décollements de rétine et la dégénérescence rétinienne liée à l’âge (fig. 4-24 et 4-26) [4, 11–13].
L’espace sous-rétinien est considéré comme un site immunologique privilégié [14] du fait de l’absence de cellule immunitaire et de la sécrétion de facteurs immunosuppresseurs par l’EPR incluant TGF-β, somastatine, thrombospondine et PEDF [15]. La persistance de cellules microgliales activées dans l’espace sous-rétinien, qui produisent des substances cytotoxiques et des cytokines, va donc altérer ce site immunologique privilégié.
Récemment, une voie transcellulaire de migration de la microglie/macrophages périvasculaires au travers de pores au niveau de l’épithélium pigmentaire a été mise en évidence par Omri et al. [16]. Ces pores permettent de faciliter, à l’état physiologique ou en cas de stress minime, le trafic de ces cellules entre la rétine et les circulations choroïdienne et systémique. Une fois passée la BHR, ni le parcours ni le rôle des cellules microgliales n’est établi de façon certaine.
Un plexus important de cellules inflammatoires et régulatrices est présent aux abords de l’EPR. Une modulation locale est donc probable, bien que dans le cas des macrophages tissulaires, la présentation d’antigènes se fasse dans les ganglions lymphatiques sous-mandibulaires ou la rate [8, 17].
Omri et al. ont également démontré que cette voie était altérée dans la rétinopathie diabétique avec la disparition de ces pores dans un modèle murin diabétique. Ainsi au stade débutant de la rétinopathie diabétique, lorsque la BHR externe est toujours intacte, la densité de ces pores augmente à la surface de l’EPR afin de réguler le processus inflammatoire par le biais de la migration de cellules microgliales et des macrophages au niveau de l’interface EPR-choroïde. À la surface de ces pores, les protéines ICAM-1 [18, 19] et caveolin-1 (CAV-1) [20], molécules impliquées dans la leukostase des cellules inflammatoires, ont été mises en évidence. La PKC-ζ qui est impliquée dans la rupture de la BHR dans l’œdème maculaire du diabète [21–23] est présente également confirmant ainsi le rôle de cette voie dans la migration des cellules inflammatoires et la genèse de l’œdème maculaire. Après 12 mois d’hyperglycémie dans ce modèle de rétinopathie diabétique, le nombre de pores transcellulaires de l’EPR et l’expression des protéines ICAM-1 et CAV-1 diminue significativement avec une accumulation de cellules microgliales/macrophages activées dans l’espace sous-rétinien.
Un tel mécanisme n’a pas été démontré dans d’autres pathologies oculaires que la rétinopathie diabétique, mais représente un mode important de migration de cellules immunitaires à travers l’endothélium vasculaire et est donc probablement ubiquitaire [24].
Que le stimulus immunologique soit systémique ou d’origine locale, les cellules microgliales une fois activées se multiplient, sont présentes en grand nombre particulièrement près des vaisseaux et expriment des marqueurs de surface typiques de cellules présentatrices pro-inflammatoires. L’expression des chimiokines CCL2, CX3C chemokine receptor 1 (CX3CR1) augmente sous l’influence de divers stress rétiniens (diabète, inflammation) [25–27]. Elles participent au recrutement de monocytes et de macrophages vers la rétine et à la sécrétion de cytokines [9, 28] telles IL-8, IL-1 activant l’inflammation, entraînant la rupture des BHR interne et externe et la dérégulation du milieu intracellulaire des CGM [29, 30–32].
On peut donc supposer que la microglie rétinienne a probablement un rôle d’immunomodulation et de tolérance au stress chronique induit par différentes pathologies rétiniennes comme la rétinopathie diabétique, l’OVCR, l’inflammation, l’uvéite : au début, il s’agit d’une régulation de la réponse inflammatoire par un échange avec d’autres cellules immunitaires par le biais de l’interface EPR–choroïde et la voie transcellulaire de l’EPR ; lorsque ces mécanismes d’immunomodulation sont dépassés, la voie transcellulaire de l’EP dysfonctionne, les cellules microgliales activées s’accumulent dans l’espace sous-rétinien activant et entretenant les mécanismes inflammatoires impliqués dans l’œdème maculaire [32].
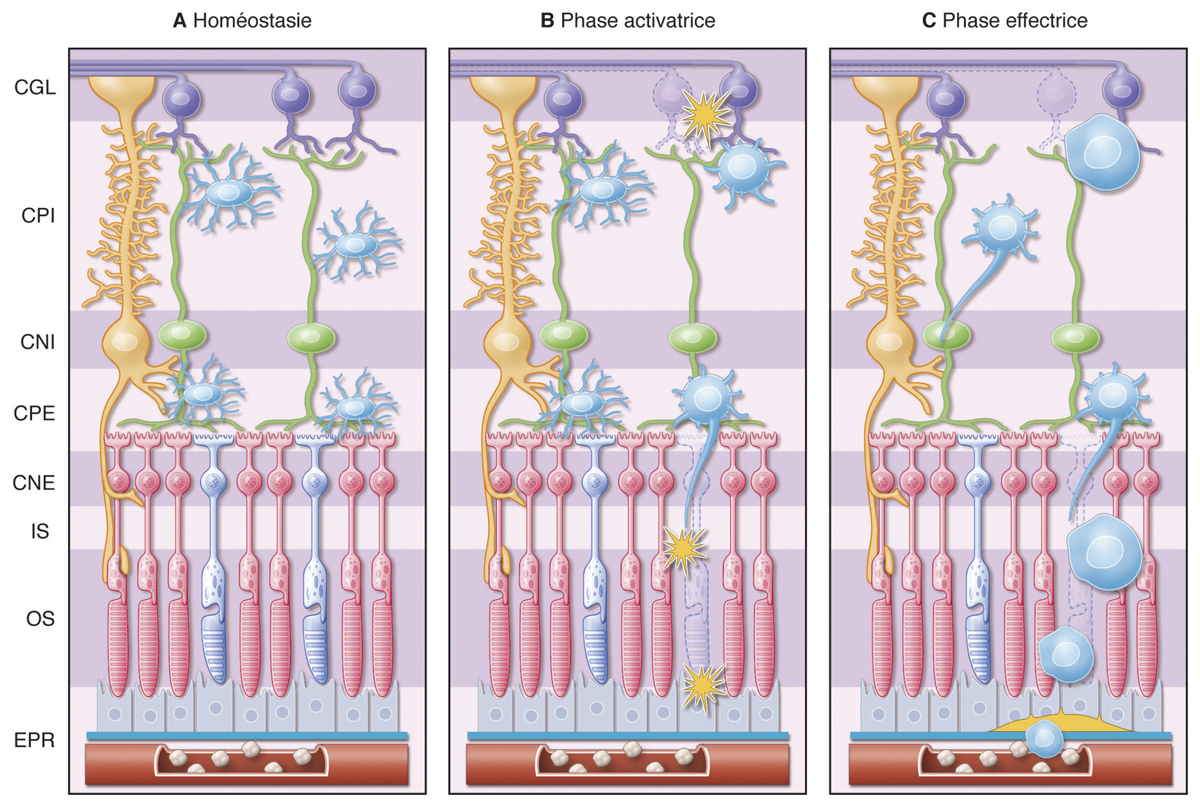
Fig. 4-24 Schéma représentatif des trois différentes phases de l’activation microgliale au niveau de la rétine.
a. À l’état physiologique, les cellules microgliales sont présentes au niveau des couches plexiformes de la rétine et ont une forme dendritique qui leur permet de sonder en permanence leur environnement grâce à de multiples prolongements cytoplasmiques. b. Différentes lésions rétiniennes amènent à une dysfonction cellulaire ou une dégénérescence des cellules de l’EPR ou des photorécepteurs ou des cellules ganglionnaires rétiniennes qui vont rapidement alerter les cellules microgliales. c. La microglie rétinienne et/ou des précurseurs dérivés de la moelle osseuse vont migrer vers le site de la lésion où ils vont prendre une forme de phagocyte amiboïde. Ces cellules effectrices peuvent être protectrices ou nocives selon leur phénotype immunologique et les cytokines produites localement. GCL : couche des cellules ganglionnaires ; CNE : couche nucléaire externe ; CNI : couche nucléaire interne ; CPE : couche plexiforme externe ; CPI : couche plexiforme interne ; EPR : épithélium pigmentaire rétinien ; IS : inner segment/segment interne ; OS : outer segment/segment externe.
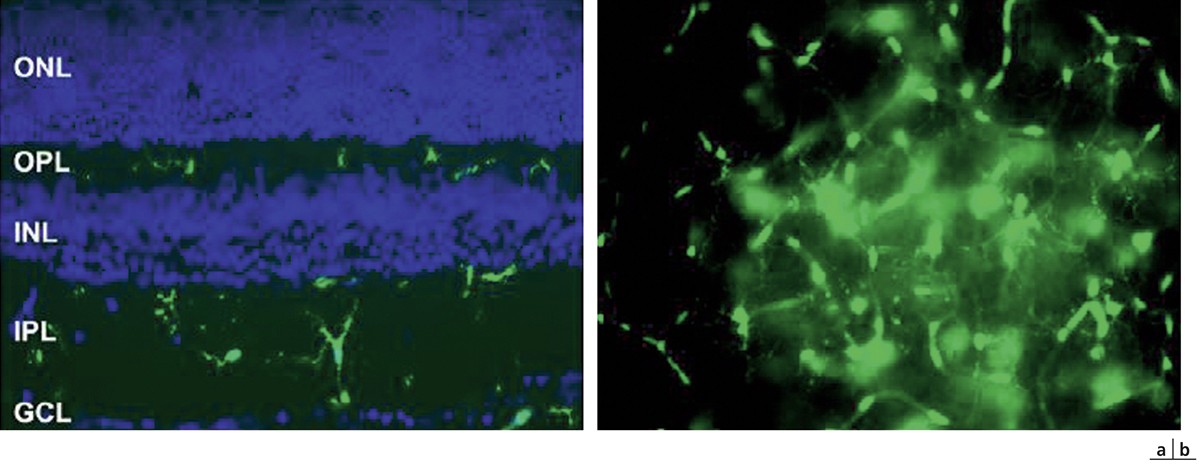
Fig. 4-25 Coupe rétinienne de souris MacGreen au microscope à fluorescence après immunomarquage EGFP+ des cellules microgliales au stade inactivé.
Vues en coupe (a) et de face (b) d’une rétine à plat au microscope à fluorescence, montrant que la microglie est exclusivement localisée au niveau des couches plexiformes. La microglie ramifiée s’étend par des prolongements cytoplasmiques arborescents. GCL : couche des cellules ganglionnaires ; INL : couche nucléaire interne ; IPL : couche plexiforme interne ; ONL : couche nucléaire externe ; OPL : couche plexiforme externe ; RPE : épithélium pigmentaire rétinien.
(Source : Karlstetter M, et al. [5]. Reproduction autorisée.)
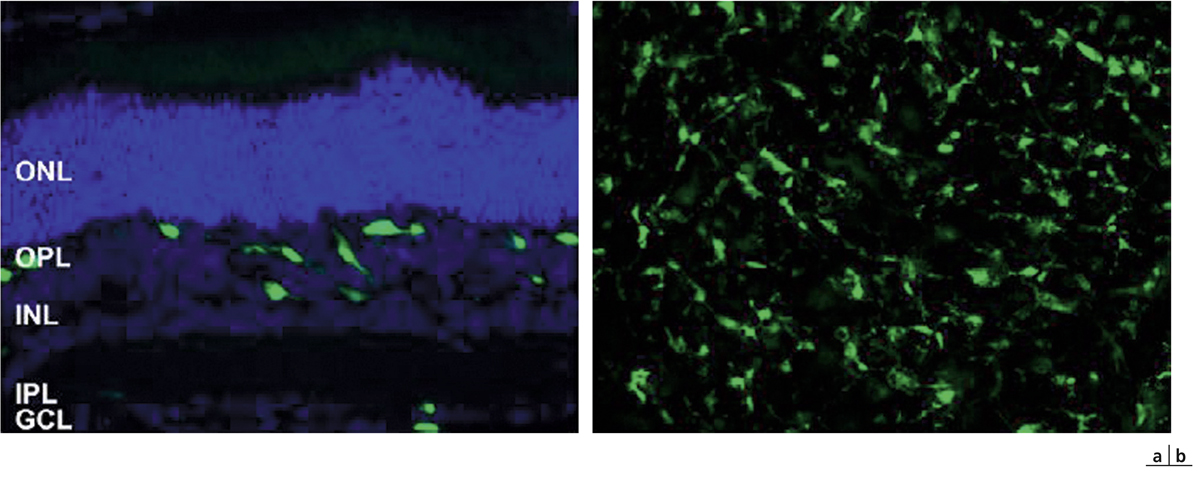
Fig. 4-26 Coupe rétinienne de souris MacGreen Rs1h-/Y présentant un modèle de dégénérescence rétinienne des photorécepteurs au microscope à fluorescence après immunomarquage EGFP+ des cellules microgliales au stade activé.
Vues en coupe (a) et de face (b) d’une rétine à plat au microscope à fluorescence. Elles montrent que la microglie activée a migré des couches plexiformes vers la couche nucléaire interne et présente une forme amiboïde. À noter l’augmentation de la densité microgliale sur la coupe de face. GCL : couche des cellules ganglionnaires ; INL : couche nucléaire interne ; IPL : couche plexiforme interne ; ONL : couche nucléaire externe ; OPL : couche plexiforme externe ; RPE : épithélium pigmentaire rétinien.
(Source : Karlstetter M, et al. [5]. Reproduction autorisée.)
La para-inflammation est une réponse tissulaire d’adaptation à un stress ou une dysfonction tissulaire se caractérisant par un état inflammatoire minime à modéré [33]. Son rôle physiologique est de restaurer la fonction et l’homéostasie tissulaires. Si ce stress devient chronique ou s’aggrave, les mécanismes d’adaptation sont dépassés et vont contribuer à l’initiation et à la progression de diverses pathologies humaines.
Au niveau ophtalmologique, la théorie de la para-inflammation a surtout été développée dans la vasculopathie diabétique, le glaucome et la dégénérescence rétinienne liée à l’âge [4, 34]. Dans la para-inflammation de la vasculopathie diabétique, les mécanismes sont similaires à ceux objectivés dans d’autres pathologies à l’origine d’œdème maculaire, mais la part inflammatoire et/ou ischémique varie selon les causes [35].
La pathophysiologie de la rétinopathie diabétique induite par l’hyperglycémie est liée à quatre voies majeures biochimiques : la voie des polyols, la voie des produits de glycation avancée, la voie de la protéine kinase C et la voie des hexosamines [36]. Toutes ces voies vont entraîner progressivement un stress chronique induisant une dysfonction rétinienne régulée et rééquilibrée par des mécanismes immunologiques, avec en particulier des liens avec les cellules de la microglie et les macrophages périvasculaires en coopération avec l’EPR.
Lorsque ces mécanismes de régulation sont dépassés, le stress oxydatif et l’inflammation entraînent la leukostase des macrophages et des cellules inflammatoires par ICAM-1 et CCL2 [27], une augmentation de la sécrétion de facteurs de croissance, de cytotoxiques et de cytokines (VEGF, angiotensine II, TNF, IL [37]) et des métalloprotéases de la matrice ; ils contribuent à la rupture de la BHR et au dysfonctionnement des photorécepteurs. Les CGM activées deviennent pro-inflammatoires et altèrent les flux ioniques et aqueux intracellulaires du fait d’un dysfonctionnement des canaux transmembranaires aqueux AQP4 et potassiques Kir4.1. L’élimination de fluide et de potassium dans la circulation générale diminue, aboutissant alors à une ballonisation des CGM (part intracellulaire de l’œdème maculaire) [38–40].
L’EPR est également altéré dans ses fonctions physiologiques et devient lui aussi pro-inflammatoire : on assiste à une diminution de la migration des cellules microgliales activées par la voie transcellulaire de l’EPR, qui s’accumulent dans l’espace sous-rétinien et entretiennent l’inflammation [16]. La rupture de la BHR externe dépasse les capacités de compensation des pompes épithéliales de l’EPR et aggrave ainsi l’accumulation d’eau dans la rétine avec l’apparition d’un décollement séreux fréquemment observé [41, 42].
L’activation microgliale et ses échanges avec les cellules immunitaires de l’interface EPR–choroïde fait partie des mécanismes immunitaires probables d’adaptation de la para-inflammation à un stress modéré de diverses pathologies rétiniennes, ceci afin que le tissu rétinien continue à fonctionner normalement.
Lorsque ces mécanismes sont dépassés, l’accumulation des cellules microgliales activées dans l’espace sous-rétinien devient inadaptée et contribue à l’entretien et l’aggravation de l’œdème maculaire par l’inflammation.
Le concept de « para-inflammation » dans le vieillissement normal et les maladies chroniques a récemment émergé et la compréhension des différentes voies para-inflammatoires est encore limitée. Les futures études portant sur les différents mécanismes médiateurs et effecteurs dans la pathogénie de la para-inflammation pourraient fournir des informations cruciales pour le développement de traitements spécifiques efficaces en particulier dans l’œdème maculaire.
[1] Stem MS, Gardner TW. Neurodegeneration in the pathogenesis of diabetic retinopathy : molecular mechanisms and therapeutic implications. Cur Med Chem 2013 ; 20 : 3241-50.
[2] Kur J, Newman EA, Chan-Ling T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog Retin Eye Res 2012 Sep ; 31 : 377-406.
[3] Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med 2012 ; 366 : 1227-39.
[4] Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009 ; 28 : 348-68.
[5] Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina : Insights from novel mouse models. Immunobiology 2010 ; 215 : 685-91.
[6] Broderick C, Duncan L, Taylor N, et al. IFN-gamma and LPS mediated IL-10 dependent suppression of retinal microglial activation. Invest Ophthalmol Vis Sci 2000 ; 41 : 2613-22
[7] Robertson MJ, Erwig LP, Liversidge J, et al. Retinal microenvrionnement controls resident and infiltrating macrohpgae function during uveoretinitis. Invest Ophthalmol vis Sci 2002 ; 43 : 2250-7.
[8] Forrester JV, Xu H, Kuffová L, et al. Dendritic cell physiology and function in the eye. Immunol Rev 2010 ; 234 : 282-304.
[9] Xu H, Chen M, Mayer EJ, et al. Turnover of resident retinal microglia in the normal adult mouse. Glia. 2007 Aug 15;55(11):1189-98. PubMed PMID: 17600341
[10] Mei C, Jiawu Z, Heping X, et al. Para-inflammation-mediated retinal recruitement of Bone Marrow derived myeloid cells following whole-body irradiation is CCL2 dependent. Glia 2012 ; 60 : 833-42.
[11] Hollborn M, Francke M, Iandiev I, et al. Early activation of inflammation- and immune response-related genes after experimental detachment of the porcine retina. Invest Ophthalmol Vis Sci 2008 ; 49 : 1262-73.
[12] Combadiere C, Feumi C, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of related macular degeneration. J Clin Invest 2007 ; 117 : 2920-8.
[13] Buschini E, Piras A, et al. Age related macular degeneration and drusen : Neuroinflammation in the retina. Prog Neurobiol 2011 ; 95 : 14-25.
[14] Jiang LQ, Jorquera M, Streilein JW, et al. Subretinal space and vitreous cavity as immunologically privileged sites for retinal allografts. Invest Ophthalmol Vis Sci 1993 ; 34 : 3347-54.
[15] Zamiri P, Sugita S, Streilein JW, et al. Immunosuppressive properties of the pigmented epithelial cells and the subretinal space. Chem Immunol Allergy 2007 ; 92 : 86-93.
[16] Omri S, Behar-Cohen F, De Kozak Y, et al. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy. Role of PKC in the goto kakizaki rat model. Am J Pathol 2011 ; 179 : 942-53.
[17] Brendecke SM, Prinz M. Do not judge a cell by its cover-diversity of CNS resident, adjoining and infiltrating myeloid cells in inflammation. Semin Immunopathol 2015 ; 37 : 591-605.
[18] McLeod DS, Lefer DJ, Merges C, et al. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol 1995 ; 147 : 642-53.
[19] Yang L, Froio RM, Sciuto TE, et al. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood 2005 ; 106 : 584-92.
[20] Millan J, Hewlett L, Glyn M, et al. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to Caveola and F-actin-rich domains. Nat Cell Biol 2006 ; 8 : 113-23.
[21] Wellner M, Maasch C, Kupprion C, et al. The proliferative effect of vascular endothelial growth factor requires protein kinase C-alpha and protein kinase C-zeta. Arterioscler Thromb vasc Biol 1999 ; 19 : 178-85.
[22] Titchenell PM, Lin CM, Keil JM, et al. Novel atypical PKC inhibitors prevent vascular endothelial growth factor-induced blood-retinal barrier dysfunction. Biochem J 2012 ; 446 : 455-67.
[23] Kozak Y, Omri B, Smith JR, et al. Protein kinase C zeta (PKC-zeta) regulates ocular inflammation and apoptosis in endotoxin-induced uveitis (EIU) : signaling molecules involved in EIU resolution by PKC-zeta inhibitor and IL 13. Am J pathol 2007 ; 170 : 1241-57.
[24] Dejana E. The transcellular railway: insights into leukocyte diapedesis. Nat Cell Biol 2006 ; 8 : 105-7.
[25] Zeng HY, Green WR, Tso MOM. Microglial activation in human diabetic retinopathy. Archives of Ophthalmology 2008 ; 126 : 227-32.
[26] Rangasamy S, McGuire PG, Franco NC, et al. Chemokine mediated monocyte trafficking into the retina : role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLOS One 2014 ; 9 : e108508.
[27] Zhang W, Rojas M, Lilly B, et al. NAD(P)H oxidase-dependent regulation of CCL2 production during retinal inflammation. Invest Ophthalmol Vis Sci 2009 ; 50 : 3033-40.
[28] Krady JK, Basu A, Allen CM, et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005 ; 54 : 1559-65.
[29] Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011 ; 91 : 461-553.
[30] Abcouwer SF. Angiogenic factors and cytokines in diabetic retinopathy. J Clin Cell Immunol 2013 ; Suppl 1(11).
[31] Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011 ; 11 : 775-87.
[32] Zeng HY, Green WR, Tso MO, et al. Microglial activation in human diabetic retinopathy. Arch ophthalmol 2008 ; 126 : 227-32.
[33] Medzhitov R. Origin and physiological roles of inflammation. Nature 2008 ; 454(7203) : 428-35.
[34] Chen M, Forrester JV, Xu H, et al. Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 Deficient Mice. PLoS ONE 2011 ; 6 : e22818.
[35] Lee WJ, Kang MH, Seong M, Cho HY. Comparison of aqueous concentrations of angiogenic and inflammatory cytokines in diabetic macular oedema and macular oedema due to branch retinal vein occlusion. Br J Ophthalmol 2012 ; 96 : 1426-30.
[36] Brownlee M. The pathobiology of diabetic complications : a unifying mechanism. Diabetes 2005 ; 54 : 1615-25.
[37] Das A, McGuire PG, Rangasamy S. Diabetic macular edema : pathophysiology and novel therapeutic targets. Ophthalmology 2015 ; 122 : 1375-94.
[38] Bringmann A, Pannicke T, Grosche J, et al. Muller cells in the healthy and diseased retina. Prog Retin Eye Res 2006 ; 25 : 397-424.
[39] Reichenbach A, Bringmann A. Immunomodulatory role of Müller cells. Müller cells in the heatlhy and diseased retina. Heidelberg : Springer ; 2010, p. 249-51.
[40] Reichenbach A, Wurm A, Pannicke T, et al. Muller cells as players in retinal degeneration and edema. Graefes Arch Clin Exp Ophthalmol 2007 ; 245 : 627-36.
[41] Peng S, Gan G, Rao VS, et al. Effects of proinflammatory cytokines on the claudin-19 rich tight junctions of human retinal pigment epithelium. Invest Ophthalmol Vis Sci 2012 ; 53 : 5016-28.
[42] Abe T, Sugano E, Saigo Y, Tamai M. Interleukin-1 beta and barrier function of retinal pigment epithelial cells (ARPE-19) : aberrant expression of junctional complex molecules. Invest Ophthalmol Vis Sci 2003 ; 44 : 4097-104.
D. Gaucher
➤ La liquéfaction du vitré est un processus physiologique favorisé par l’âge, dont la conséquence est le décollement progressif du cortex vitréen postérieur.
➤ Certaines conditions pathologiques comme le diabète ou l’inflammation chronique sont à l’origine d’une liquéfaction précoce du vitré, associée à des adhérences vitréorétiniennes renforcées, conduisant fréquemment à un vitréoschisis, sans décollement de la hyaloïde postérieure.
➤ Ce décollement postérieur du vitré pathologique est à l’origine de tractions chroniques pouvant induire ou entretenir un OM.
➤ Le cortex vitréen postérieur constitue un réservoir de cytokines, notamment des cytokines pro-inflammatoires au contact de la rétine, favorisant l’OM.
Les modifications du vitré sont associées à nombreuses pathologies maculaires. Cette relation entre vitré et rétine maculaire a été remarquée dès les années 1960. Les travaux de Charles Schepens puis de Jerry Sebag ont probablement été à la base de la compréhension de cette relation, en étudiant les structures histologiques du vitré. Depuis les années 1990 et surtout 2000, la tomographie à cohérence optique (OCT) a véritablement révolutionné l’analyse de l’interface vitréomaculaire et a permis d’expliquer et de découvrir de nombreuses pathologies liées à cette interface. Malgré ces progrès, les phénomènes d’adhésion, de contraction et de séparation du vitré restent assez mal connus. Il n’en reste pas moins que ces derniers jouent un rôle dans la survenue ou l’entretien de certains OM.
Le vitré est une substance gélatineuse qui contient 98 % d’eau, quelques cellules phagocytaires et des hyalocytes qui entretiennent un réseau de glysaminoglycanes (GAG) de type acide hyaluronique, héparane sulfate ou chondroïtine sulfate, en relation avec une structure de fibrilles de collagène de type II.
Les GAG sont des éléments fondamentaux de la matrice extracellulaire et du vitré. Ce sont de longues chaînes composées d’une répétition d’unités dissacharidiques (fig. 4-27). Les GAG forment un maillage d’éléments carbohydratés, contenant beaucoup d’eau. Cette caractéristique explique la consistance gélatineuse du vitré, sa résistance à la compression et sa capacité à maintenir une pression oculaire. La viscosité du vitré est 4 fois supérieure à celle de l’eau mais son indice de réfraction de 1,336 est très similaire. Enfin, à l’exception de l’acide hyaluronique, les GAG se lient de façon non covalente à des protéines et forment des « protéoglycanes ». Ces protéoglycanes permettent la liaison du vitré avec les cellules de Müller par le biais de protéines membranaires et des fibres de collagène de la matrice extracellulaire (fig. 4-27).
Les fibrilles de collagène sont des structures protéiques hélicoïdales résistantes aux forces de tension. Elles sont « empilées » et condensées en regard de la rétine et sont adhérentes à la limitante interne de la rétine que constitue la membrane basale des cellules de Müller. Elles peuvent avoir des liaisons cellulaires par l’intermédiaire direct de la molécule d’adhérence « intégrine » ou par le biais de protéines d’adhésion comme la laminine, la fibronectine ou, comme dit précédemment, par le biais des GAG (fig. 4-27).
Fig. 4-27 Schéma représentant l’interface vitréorétinienne.
Les protéines membranaires de type « intégrine » se lient au collagène de la limitante interne et du vitré directement ou par l’intermédiaire de protéines d’adhésion comme la laminine, ou encore grâce aux glycosaminoglycanes (acide hyaluronique notamment) qui se lient aussi bien à l’intégrine qu’aux fibrilles de collagène.
La limitante interne est épaissie au niveau du clivus (autour de la fovéola) et s’amincit vers la base du vitré en avant et vers la fovéola en arrière. Elle est plus fermement adhérente aux cellules de Müller grâce à des plaques d’attachement ou hémidesmosomes, constitués d’intégrines liant le cytosquelette des cellules de Müller à la matrice extracellulaire de la limitante interne en moyenne et extrême périphérie rétinienne.
La limitante interne est, comme le vitré, constituée de glycoprotéines et de fibrilles de collagènes de plusieurs types (IV, VI et XVIII). Le collagène de type XVIII se lie par des liaisons non covalentes aux protéines de la matrice extracellulaire. L’opticine est l’une d’entre elles. Elle se lie à plusieurs protéoglycanes dont la chondroïtine sulfate présente dans le cortex vitréen, elle-même liée aux fibrilles de collagène du vitré. Opticine et chondroïtine sulfate semblent être deux protéines importantes dans l’adhésion vitréorétinienne.
L’adhérence du vitré à la rétine varie d’un point de vue anatomique. L’adhérence est plus forte dans les zones où la limitante interne est la plus fine : base du vitré, papille, fovéola et en regard des vaisseaux rétiniens. Elle varie aussi en fonction de l’âge, puisque l’adhérence diminue avec le temps en raison des modifications des caractéristiques physiques du vitré avec l’âge. Au cours du vieillissement, le stress oxydatif modifie la structure de l’acide hyaluronique. Le collagène pourrait être aussi atteint par le biais d’une lyse enzymatique engendrée par les métalloprotéinases. On note une liquéfaction progressive du vitré [1], probablement due à une dissociation des fibres de collagène et de l’acide hyaluronique. Cette dissociation est caractéristique en avant de la fovéa. Kishi et al. ont décrit, depuis longtemps déjà, la présence d’une cavité dans le cortex vitréen en avant de la rétine maculaire appelée « poche prémaculaire ». Elle pourrait causer une adhérence particulière du cortex vitréen à la rétine en arrière de la poche et se voit fréquemment dans les trous maculaires (fig. 4-28) [2].
Une des conséquences de la liquéfaction du vitré est le décollement progressif du cortex vitréen postérieur de la limitante interne. Une étude post mortem a montré que plus le vitré était liquéfié, plus il était décollé totalement du pôle postérieur [3]. Grâce à l’OCT-3, Uchino et al. [4] ont démontré l’évolution progressive du décollement postérieur du vitré (DPV) comme cela a été confirmé en spectral-domain optical coherence tomography (SD-OCT) récemment [5] : le décollement du cortex postérieur débute souvent en périfovéolaire en nasal puis en temporal de la fovéola. Il est probable que des poches de liquéfaction provoquent des ruptures localisées du cortex et permettent au fluide de passer dans l’espace prérétinien, entraînant par le biais de protéases une lyse des complexes de protéoglycanes et des fibres de collagène. La progression du fluide provoquerait l’extension du DPV selon une chronologie et une topologie typiques (fig. 4-29) [4, 5]. En effet, le décollement de la hyaloïde postérieure est d’abord paramaculaire (fig. 4-29a), puis la hyaloïde se décolle en progressant vers la fovéola au stade du DPV parafovéolaire (fig. 4-29b). Le DPV s’étend encore vers le centre fovéolaire, laissant une attache centrofovéolaire (fig. 4-29c). Le DPV s’étend ensuite vers la périphérie supérieure et temporale et enfin vers la périphérie nasale et inférieure. L’attache fovéolaire lâche au stade de DPV avec attache papillaire (fig. 4-29d). Le DPV est considéré comme complet lorsque l’attache papillaire cède. Le processus de DPV s’étale sur plusieurs mois ou années. Classiquement, le DPV survient après 50–60 ans, 65 % des patients au-delà de 65 ans auraient un DPV. Si l’on s’intéresse aux stades du DPV, on remarque que le décollement parafovéolaire débute dans 30 % des cas dès 30–40 ans, mais entre 60 et 70 ans, seuls 70 % des patients ont un DPV complet [5].
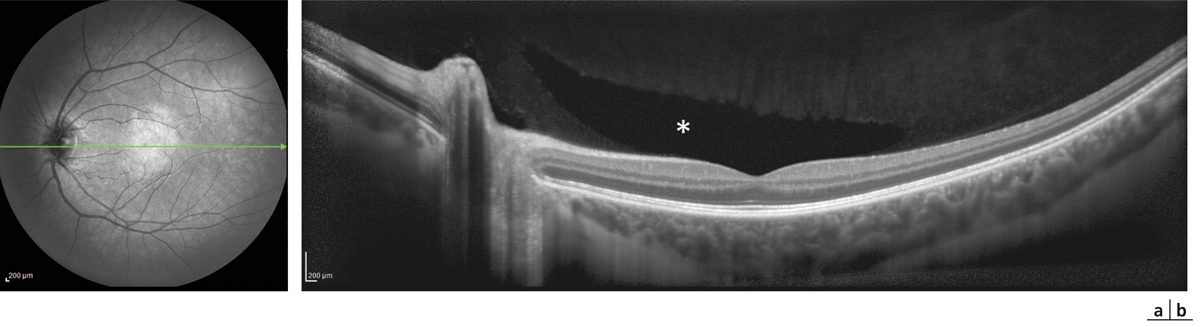
Fig. 4-28 Poche prémaculaire vue en OCT 55° (Spectralis®, Heidelberg, Allemagne).
Le vitré liquéfié de la poche prémaculaire est très visible (*) chez cette jeune patiente de 30 ans. Cet espace est hyporéflectif en comparaison avec le vitré fibrillaire non liquéfié environnant.
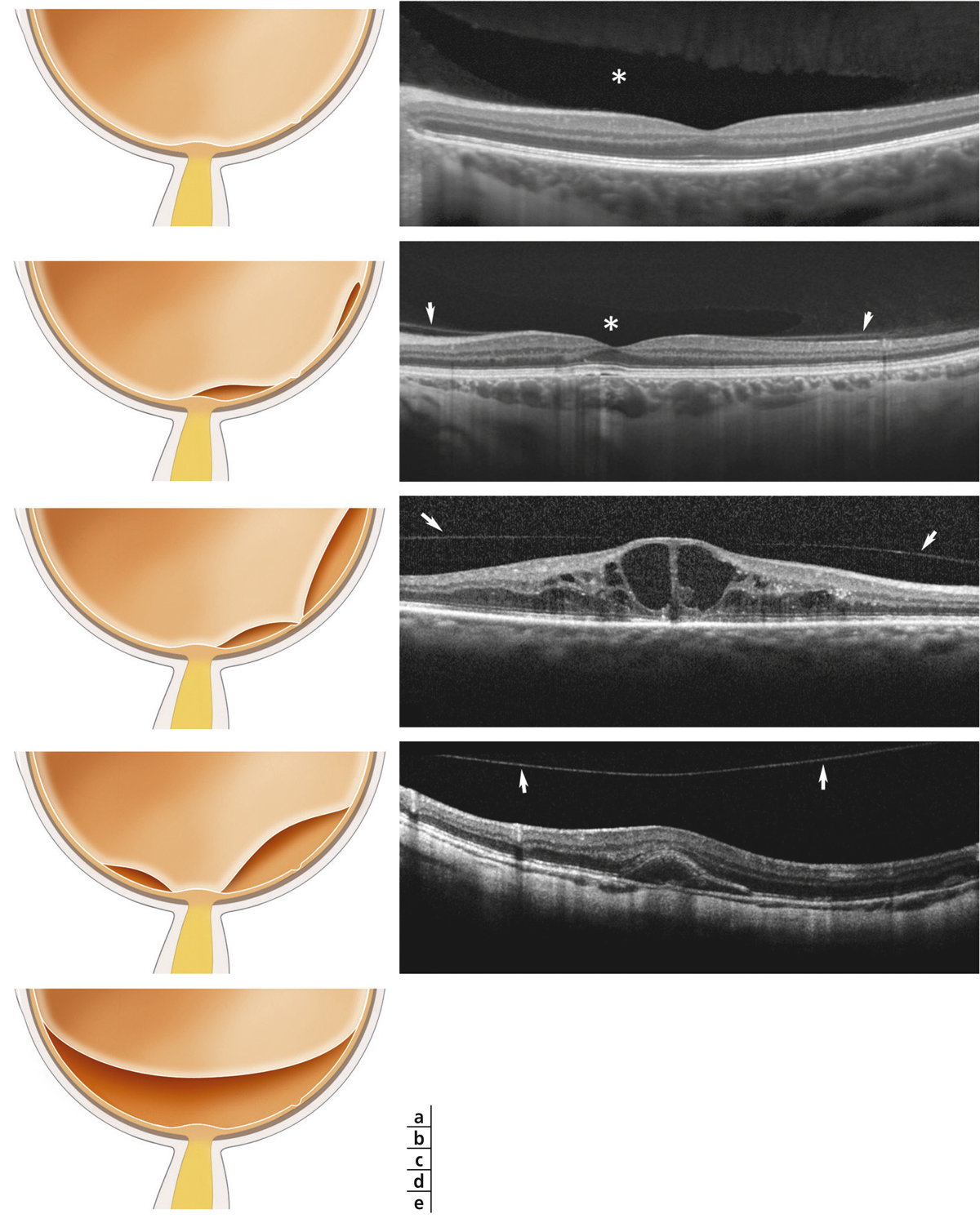
Fig. 4-29 Schéma représentant les différents stades de DPV et exemples OCT correspondant en situation clinique.
a. Absence de DPV, la hyaloïde postérieure du vitré est partout adhérente à la rétine. Sur l’exemple OCT, la poche prémaculaire (*) ne correspond pas à un DPV, car on ne visualise pas la hyaloïde hyper-réflective. b. DPV paramaculaire, la hyaloïde se décolle peu de la rétine de part et d’autre du centre fovéolaire (flèches) mais l’attache centrale reste large en regard d’une bourse prémaculaire (*). c. DPV périfovéolaire, l’attache centrofovéolaire est limitée à la fovéola comme dans l’exemple, ou le DPV est associé à un œdème maculaire diabétique. d. DPV avec attache papillaire, la hyaloïde est détachée de tout le pôle postérieur à l’exception du nerf optique. Elle est visible en OCT en avant de la rétine, dans un cas de rupture de la membrane de Bruch avec hémorragie maculaire. e. DPV complet, l’adhérence de la hyaloïde est uniquement antérieure au niveau de la base du vitré, la hyaloïde n’est plus visible en OCT.
À part l’âge, certaines conditions modifient l’apparition du DPV, notamment le diabète, la myopie forte et les maladies oculaires inflammatoires ou encore les maladies du tissu conjonctif (Marfan, Elher-Danlos, etc.). Dans ces conditions, la structure du vitré est probablement modifiée, la liquéfaction du vitré survient plus tôt mais l’adhérence vitréorétinienne y est plus forte, ce qui conduit fréquemment à un vitréoschisis, c’est-à-dire des lacunes de liquéfaction intravitréenne sans décollement de la hyaloïde postérieure.
La traction du vitré ou de membranes prémaculaires sur la rétine est responsable d’épaississement rétinien dans plusieurs pathologies. Dès 1966, Jaffé et al. [6, 7] ont noté que des tractions antéropostérieures dues à un décollement partiel du vitré pouvaient être associées à une baisse de vision. En 1984, Schepens et Sebag ont montré que le vitré était adhérent aux cellules de Müller via la limitante interne et ont suspecté le rôle de cette adhérence dans la pathogénie de l’OM. La traction rétinienne chronique est associée dans 60 à 85 % des cas à des kystes ou à des décollements séreux rétiniens, témoignant d’une rupture de la BHR interne. Inversement, les OM chroniques induisent la formation de membranes prémaculaires ayant un rôle de traction sur la rétine [8]. Quelques études anciennes ont montré un lien entre le décollement partiel du vitré et l’œdème maculaire dans l’occlusion veineuse [9, 10]. Ce rôle aggravant du vitré est également décrit dans les œdèmes de la DMLA exsudative [11, 12] et dans le diabète [13]. De Smet a montré dans une méta-analyse récente que le risque d’avoir un œdème maculaire (toutes pathologies confondues : rétinopathie diabétique, occlusion veineuse, etc.) était diminué en cas de DPV, et que le risque de persistance de l’OM était associé à l’absence de DPV complet [14]. Ces observations montrent que l’adhérence du vitré prolongée sur le centre fovéolaire peut donc aggraver un OM. Par ailleurs, elle représente dans certaines pathologies comme le rétinoschisis du myope fort ou le syndrome de traction maculaire, la cause principale de l’OM (fig. 4-30).
D’autres rôles aggravants ont été attribués au vitré dans la physiopathologie de l’OM : d’une part, un rôle de réservoir de cytokines et notamment des cytokines pro-inflammatoires dont le VEGF, qui resteraient ainsi au contact de la rétine et d’autre part, un rôle de barrière à l’oxygénation. Ces deux facteurs, encore discutés, favoriseraient la survenue ou l’aggravation de la rupture de la BHR qui alimente l’OM [15].
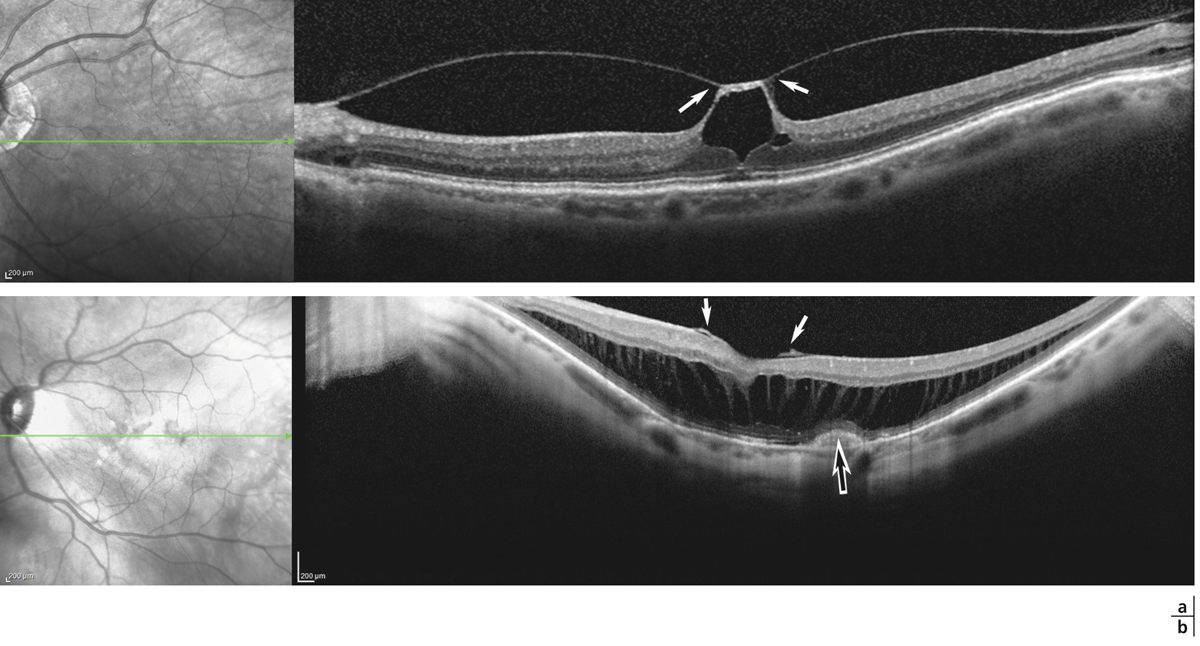
Fig. 4-30 Exemples d’épaississement maculaire dû majoritairement à la traction du vitré.
a. Syndrome de traction vitréorétinienne, l’attache fovéolaire du vitré ne cède pas (flèches) et entraîne un déplacement antérieur de la fovéola et la formation d’un kyste fovéolaire. b. Fovéoschisis du myope fort, l’attache vitréenne est complète ; on devine la hyaloïde épaissie au bord de la fovéola (flèches). On note également un probable néovaisseau choroïdien myopique (flèche creuse). Dans ces deux types d’« œdèmes » maculaires, la suppression de l’adhérence vitréorétinienne par voie de vitrectomie suffit souvent à restaurer en partie l’anatomie et la fonction rétiniennes.
Le vitré est un tissu qui évolue dans le temps. Les modifications structurelles du vitré influencent l’adhésion entre ce dernier et la rétine. Le DPV est un phénomène naturel progressif lié à ces modifications. La classification récente définissant les différentes étapes du DPV et son éventuelle évolution pathologique a le mérite de définir plus clairement la terminologie des différentes situations. Dans certaines conditions, l’absence de DPV ou le DPV partiel semblent favoriser la survenue et l’entretien de l’OM. Le vitré est à ce titre une cible thérapeutique importante dans le traitement de certains OM.
[1] Sebag J. Age-related changes in human vitreous structure. Graefes Arch Clin Exp Ophthalmol 1987 ; 225 : 89-93.
[2] Kishi S, Hagimura N, Shimizu K. The role of the premacular liquefied pocket and premacular vitreous cortex in idiopathic macular hole development. Am J Ophthalmol 1996 ; 122 : 622-8.
[3] Larsson L, Osterlin S. Posterior vitreous detachment. A combined clinical and physiochemical study. Graefes Arch Clin Exp Ophthalmol 1985 ; 223 : 92-5.
[4] Uchino E, Uemura A, Ohba N. Initial stages of posterior vitreous detachment in healthy eyes of older persons evaluated by optical coherence tomography. Arch Ophthalmol 2001 ; 119 : 1475-9.
[5] Itakura H, Kishi S. Evolution of vitreomacular detachment in healthy subjects. JAMA Ophthalmol 2013 ; 131 : 1348-52.
[6] Jaffe NS, Light DS. Vitreous changes produced by cataract surgery. A study of 1,058 aphakic eyes. Arch Ophthalmol 1966 ; 76 : 541-53.
[7] Jaffe NS. Vitreous traction at the posterior pole of the fundus due to alterations in the vitreous posterior. Trans Am Acad Ophthalmol Otolaryngol 1967 ; 71 : 642-52.
[8] Reese AB, Jones IS, Cooper WC. Macular changes secondary to vitreous traction. Am J Ophthalmol 1967 ; 64 : 544-9.
[9] Trempe CL, Takahashi M, Topilow HW. Vitreous changes in retinal branch vein occlusion. Ophthalmology 1981 ; 88 : 681-7.
[10] Kado M, Jalkh AE, Yoshida A, et al. Vitreous changes and macular edema in central retinal vein occlusion. Ophthalmic Surg 1990 ; 21 : 544-9.
[11] Simpson AR, Petrarca R, Jackson TL. Vitreomacular adhesion and neovascular age-related macular degeneration. Surv Ophthalmol 2012 ; 57 : 498-509.
[12] Jackson TL, Nicod E, Angelis A, et al. Vitreous attachment in age-related macular degeneration, diabetic macular edema, and retinal vein occlusion : a systematic review and metaanalysis. Retina 2013 ; 33 : 1099-108.
[13] Gaucher D, Tadayoni R, Erginay A, et al. Optical coherence tomography assessment of the vitreoretinal relationship in diabetic macular edema. Am J Ophthalmol 2005 ; 139 : 807-13.
[14] de Smet MD, Gad Elkareem AM, Zwinderman AH. The vitreous, the retinal interface in ocular health and disease. Ophthalmologica 2013 ; 230 : 165-78.
[15] Stefansson E, Loftsson T. The Stokes-Einstein equation and the physiological effects of vitreous surgery. Acta Ophthalmol Scand 2006 ; 84 : 718-9.
M. Streho
➤ Il n’existe pas toujours une bonne corrélation entre les évaluations fonctionnelles et anatomiques.
➤ Les meilleures échelles d’acuité visuelle (AV) sont les échelles logarithmiques qui assurent une discrimination homogène des items pour chaque ligne d’AV et une variation régulière de la taille des items d’une ligne à l’autre.
➤ Il existe une faible corrélation entre les différentes échelles d’AV, notamment entre l’échelle de Snellen (couramment utilisée en pratique clinique) et l’échelle Early Treatment Diabetic Retinopathy Study (ETDRS).
➤ Il existe une faible corrélation entre l’AV et l’épaisseur maculaire dans les OM. L’OCT est donc un paramètre important mais ne remplace pas la mesure d’AV pour évaluer la fonction visuelle.
L’OM est défini par une accumulation de liquide et autres substances (lipides, protides) entraînant un épaississement du tissu rétinien et une diminution de la fonction rétinienne. Les causes sont multiples (diabète, uvéite, occlusion veineuse, DMLA, etc.), mais les mécanismes sont principalement liés à un dysfonctionnement de la BHR et une accumulation des facteurs inflammatoires dont le taux de VEGF élevé [1, 2]. Le diagnostic peut être clinique et indirect en mesurant l’acuité visuelle ou clinique et direct par examen du fond d’œil. Le diagnostic peut être paraclinique à l’aide de l’angiographie à la fluorescéine et/ou de l’OCT [3, 4]. On distingue principalement les tests fonctionnels, testant la fonction visuelle et donc le retentissement de l’OM, des tests anatomiques ou structurels, visualisant directement la présence et la quantification de l’œdème au sein de la macula. Il n’existe pas toujours une bonne corrélation entre les évaluations fonctionnelles et anatomiques, comme dans le glaucome, où la corrélation entre les examens fonctionnels (champ visuel) et anatomiques (OCT papillaire) n’est pas bonne ou parfois décalée dans le temps. L’objectif des évaluations est de dépister l’OM au plus tôt pour pouvoir le traiter et limiter son retentissement fonctionnel. En effet, n’oublions pas que le patient se plaint de gêne fonctionnelle et le praticien traite une atteinte anatomique. Il est donc primordial de connaître les différentes techniques d’évaluation de la fonction et de la structure visuelle dans l’OM pour faire converger les attentes du patient avec les possibilités du praticien.
La mesure de l’acuité visuelle (AV) est la base de toute démarche diagnostique et thérapeutique en ophtalmologie, l’objectif du traitement étant d’améliorer voire de restaurer l’AV du patient. Chez l’enfant, l’AV reflète la maturation visuelle qui s’opère dès la naissance. Sa mesure est variable et permet le dépistage de l’amblyopie, conditionnant l’acquisition de la lecture et des orientations scolaires. Son évaluation précoce est déterminante pour proposer une rééducation.
La principale définition de l’AV est la capacité à discriminer deux points distincts (résolution de notre œil comme celui d’un appareil OCT). Il s’agit du « minimum séparable » ou encore acuité angulaire, représentant l’angle sous lequel est vu le plus petit écart distinguant deux points et sous-tendu par l’anatomie des cônes fovéolaires (expérimentalement 36 secondes d’arc correspondant à 3 µm d’écart entre deux cônes). La minute d’arc a été adoptée comme référence de la normalité [6]. L’acuité visuelle peut toutefois être définie par d’autres notions (voir chapitre 5.1).
Il existe plusieurs types d’échelles d’AV : décimale (Monoyer) ou géométrique (logarithmique). Le test idéal doit être simple, rapide, sensible, précis, fiable et reproductible. Les tests les plus fiables sont basés sur l’acuité angulaire [7]. Les échelles de mesure morphoscopique, étalonnées en comparant leurs résultats à ceux des échelles angulaires, sont basées sur la reconnaissance de la forme d’optotype (dessins, chiffres, lettres, etc.). Si elles suivent une progression arithmétique (échelle décimale dite de Monoyer), il y a autant de différence entre 1/10 et 2/10 qu’entre 5/10 et 10/10. Les progrès dans les hautes valeurs d’AV y sont donc surestimés, au contraire des valeurs basses, car l’échelle offre beaucoup moins de niveaux d’AV en deçà de 3/10 (tableau 4-4). Idéalement, la présentation des échelles doit donc suivre une progression géométrique. Celle-ci assure une discrimination homogène des items pour chaque ligne d’AV et une variation régulière de la taille des items d’une ligne à l’autre. Cela suppose un même nombre d’items par ligne, un espace constant entre chaque item, un espace entre une ligne et la suivante égal à la taille des items de la ligne suivante. C’est tout l’intérêt des échelles logarithmiques comme l’échelle ETDRS. Le plus souvent, elles progressent de 26 %. La constance du nombre égal d’optotypes par ligne offre aussi la possibilité d’accorder le même sens à une erreur tout au long de l’échelle (0,02 unité) (tableau 4-5). D’autres facteurs interviennent en plus de la dimension angulaire :
-
la luminance de fond (150 à 650 cd/m2) et de l’environnement (10 à 25 % de celle du fond, en général un éclairage artificiel de ≈ 100 lux) ;
-
le contraste (idéalement 100 % car la diffraction fait qu’il n’est plus que de 20 % au niveau de la rétine) ;
-
la couleur (l’AV est maximale pour une lumière blanche, l’œil n’étant emmétrope que pour le jaune-vert et le pigment des cônes fovéaux surtout sensible à 555 nm) ;
-
la forme du test (les échelles morphoscopiques font appel à des processus cognitifs en plus du fonctionnement des cônes ;
-
la facilité de reconnaissance des chiffres et lettres est variable ainsi le « I » est le plus facile, le « B » le plus difficile ;
-
la distance de présentation (4 m pour la vision de loin, 1 m pour la basse vision et 40 cm pour la vision de près) ;
-
le temps de présentation (optimal avec 1 seconde, sa réduction diminue l’AV). Tout cela suppose un contrôle régulier de la qualité des projecteurs mais également des conditions de réalisation de la mesure. Les principales règles à respecter sont :
1.vérifier la réfraction (et la qualité du centrage des verres),
2. s’assurer de la distance de présentation,
3. éviter les lettres qui se confondent,
4. renouveler régulièrement les échelles papier,
5. préférer une échelle géométrique d’au moins 5 optotypes par ligne,
6. convertir les résultats des échelles décimales en données logarithmiques,
7. recourir aux échelles de l’enfant en cas d’obstacle linguistique ou d’illettrisme,
8. disposer d’échelles pour malvoyant pour les AV < 5/10 (encadré 4-1
Encadré 4-1 - Règles de conversion des échelles décimales en logarithmiques• LogMAR = Log 1/AV décimale
• LogMAR = Log de base 10 (angle de vision en minutes d’arc)
Score ETDRS = 85–50 LogMAR
).
D’autres mesures de la fonction visuelle sont également importantes, notamment la sensibilité au contraste et le champ visuel central. Trois patients avec 20/40 d’acuité peuvent très bien avoir des perceptions différentes du contraste ou du champ visuel central et avoir ainsi des performances de lecture très différentes. Pour certains patients, d’autres mesures telles que l’étude du champ visuel périphérique seront également importantes [8].
Tableau 4-4 Comparaison de la progression des échelles.
Tableau 4-5 Avantages et inconvénients des échelles arithmétiques et logarithmiques de l’adulte.
La capacité de discerner le contraste est une fonction visuelle distincte de l’acuité. Des tests formels explorant la sensibilité au contraste utilisent notamment les diagrammes de Vistech, qui permettent de tracer la courbe de sensibilité au contraste sur une large plage de fréquence spatiale, ou le diagramme de Pelli-Robson qui présente de grands optotypes sur une large plage de contrastes. Bien que l’acuité et la sensibilité au contraste soient souvent corrélées, elles peuvent cependant se dissocier dans certaines pathologies (neuropathies optiques, atrophies localisées). Les patients ayant une faible sensibilité au contraste présentent des difficultés à voir le bord des marches, à lire les imprimés de couleur claire, à conduire dans des conditions météorologiques difficiles telles que le brouillard ou la neige (voir chapitre 5.2).
Le test de la grille d’Amsler peut sous-estimer la perte de champ central en raison d’un phénomène de compensation ou de « remplissage » physiologique.
La procédure la mieux adaptée à l’évaluation du champ visuel central est d’effectuer une micropérimétrie maculaire qui va chercher l’emplacement du fond d’œil et détecter ensuite la direction que prend le regard du patient lorsqu’une cible lui est présentée. La micropérimétrie détermine quel est le point de la rétine qui fixe la cible (avec la qualité de la fixation pour certains appareils), la présence de scotome et la relation point de fixation/scotomes. La plupart des patients atteints d’un scotome central développent une pseudo-fovéa fonctionnelle excentrique, appelée preferred retinal locus (PRL). Connaître l’emplacement du PRL par rapport au scotome peut aider à comprendre les difficultés du patient. Les scotomes peuvent varier en taille, en forme, en nombre et densité, et peuvent ne pas correspondre à l’apparition d’atrophie au fond d’œil. Il est très important de tenir compte de ce manque de correspondance (voir chapitre 5.2).
La façon la plus simple d’évaluer l’éblouissement est de retracer l’histoire clinique. Cependant les cliniciens peuvent également tester l’AV et la sensibilité aux contrastes avec et sans source de lumière, placée à proximité de la cible et dirigée vers le patient. Le Brightness Acuity Tester est un appareil de poche qui permet au patient de visualiser une cible éloignée à travers une petite coupole qui surcharge les yeux. Les éblouissements sont source d’inconforts ou de troubles de la vision causés par la lumière diffusée. Les patients percevant mal les contrastes ont souvent besoin d’un éclairage amplifié, ce qui peut aggraver l’éblouissement.
Les troubles acquis de vision des couleurs rendent les tâches nécessitant une identification de couleurs plus difficiles. Les tables pseudo-isochromatiques couramment utilisées (plaque d’Ishihara) ne permettent pas une bonne évaluation des atteintes de la reconnaissance du bleu-jaune. Elles détectent plutôt, de manière générale, les déficits héréditaires de la vision rouge-vert. Il existe donc des tests permettant de détecter aussi bien les déficits de vision du bleu-jaune que les déficits du rouge-vert, comme les plaques de Hardy-Rand-Rittler et le pannel D15 de Farnsworth (voir chapitre 5.2).
En plus de l’appréciation de la fonction visuelle, l’évaluation globale du patient doit inclure ses capacités d’exécution des tâches nécessitant la vue. Certaines activités peuvent être étudiées comme la lecture (vitesse, distance, taille des caractères, etc.). Globalement, il existe des questionnaires de qualité de vie (et de vue) validés dans différentes pathologies comme le Visual Function Questionnaire 25 (VFQ-25) dans la DMLA, cataracte, glaucome, rétinopathie diabétique, kératocône, décollement de rétine, trou maculaire, etc. Le questionnaire comporte 25 critères avec un score de 0 à 100 comportant plusieurs items : vision générale, activité de près/de loin, vision périphérique, vision des couleurs dans les activités de la vie courante (lecture, conduite, etc.) (voir chapitre 5.2).
L’œdème maculaire s’explore également par des examens anatomiques dont historiquement le fond d’œil, l’angiographie à la fluorescéine et plus récemment l’OCT ou encore l’angio-OCT. Ces examens permettent de visualiser directement la présence ou non d’OM, mais également une quantification objective de cet œdème. L’OCT a été développé pour mesurer de manière objective, précise (5 à 10 µm), reproductible et fiable l’épaisseur maculaire. Parmi les mesures, nous utilisons principalement l’épaisseur maculaire centrale, mais également le volume maculaire. Il est important de vérifier l’absence d’artefacts dans la segmentation pour avoir une mesure d’épaisseur fiable. Chaque appareil possède un algorithme propre quant à la mesure des épaisseurs maculaires. Une différence existe pour la normale entre ces différents appareils. Ces techniques sont plus longuement développées dans le chapitre 5.2.
Plusieurs études ont montré la faible corrélation entre les différentes échelles d’AV, notamment entre l’échelle de Snellen (couramment utilisée en pratique clinique) et l’échelle ETDRS. Kaiser a montré dans une étude prospective portant sur 163 yeux en pratique courante la faible corrélation entre les deux échelles d’AV [9]. Dans son étude, l’AV moyenne est : en échelle de Snellen de 0,67 logMAR (20/94), en échelle ETDRS à 4 m de 0,57 logMAR (20/69), en ETDRS 2 m de 0,51 logMAR (20/65), ce qui fait une différence moyenne entre les différentes échelles de 6,5 lettres ( p < 0,000000001). Il a montré que quand l’AV baisse, la variabilité entre les différentes échelles se majore. Les études de sous-groupes ont montré que la différence se majorait en cas de faible AV (< 20,200) allant jusqu’à 10 lettres ( p < 0,0000002). Il existe par ailleurs des différences en fonction des pathologies avec une plus grande discordance en cas de DMLA néovasculaire mais également en présence de DMLA atrophique ou d’œdème maculaire diabétique (OMD). Falkenstein et al. ont montré la faible corrélation entre ces deux échelles d’AV dans la DMLA [10]. En effet, dans une étude cross-sectionnelle portant sur 190 yeux dans un groupe atteint de DMLA, l’AV moyenne était en échelle de Snellen de 0,78 logMAR (20/120) et en échelle ETDRS de 0,54 logMAR (20 /70, p < 0,001). La différence entre les deux échelles est maximale (> 3 lignes) en cas de faible AV (< 20/200) et minimale (< 1 ligne) en cas de bonne AV (> 20/30). L’échelle ETDRS semble être la plus adaptée aux faibles AV (< 20/200) correspondant à des DMLA (ou autres maculopathies) avancées. Ces constatations doivent être prises en compte pour interpréter avec précaution les résultats de gain d’AV dans les études pivotales (gain d’AV exprimé en échelle ETDRS) et les études de la vraie vie (gain d’AV souvent exprimé en échelle Snellen). Une étude réalisée à l’hôpital Lariboisière (en cours de publication) a montré une relative bonne corrélation entre les deux échelles (Snellen et ETDRS) dans les œdèmes maculaires diabétiques avec une AV relativement conservée. Il s’agit d’une étude prospective, non interventionnelle ayant porté sur 62 yeux. L’AV moyenne était de 0,5 logMAR (20/63) en échelle de Snellen et 0,506 logMAR (20/64) en échelle ETDRS. Cette étude montre une bonne corrélation avec un facteur de corrélation de 0,939 ( p < 0,0001).
Toutes les études portant sur les échelles d’AV montrent les limites de l’échelle de Snellen avec une faible fiabilité, une grande variabilité d’un examen à l’autre (en moyenne ± 0,29 à 0,33 logMAR) [11, 12]. Ainsi depuis 1993, l’échelle ETDRS est devenue le gold standard avec de bonnes précision, fiabilité et reproductibilité pour les hautes mais également les faibles AV [13]. La variabilité de l’échelle ETDRS est de ± 0,07 à 0,16 logMAR pour les sujets sains et ± 0,15 à 0,2 logMAR pour les sujets atteints de maculopathies [14]. La principale limite de la mesure en échelle ETDRS est la durée de l’examen (2 fois plus longue que l’échelle de Snellen).
La présence d’un OMD (ou autre) est associée à une réduction de l’AV même si celle-ci est variable. De plus, l’apparition de traitements (laser, anti-VEGF, corticoïdes, etc.) entraînant une diminution de l’OM a pour effet une amélioration de l’AV. L’OCT permet de mesurer de manière objective, fiable, précise et reproductible l’épaisseur maculaire. Plusieurs études se sont intéressées à la corrélation entre les mesures de l’épaisseur maculaire en OCT et l’AV. La corrélation semble relativement faible et varie globalement de 0,28 à 0,73 [3]. Si cette corrélation était suffisamment forte, l’OCT serait, à lui seul, suffisant pour évaluer la fonction visuelle. Toutefois, les études montrent bien la nécessité d’associer les mesures d’AV aux mesures d’OCT. En effet, une étude du Diabetic Retinopathy Clinical Research Network s’est intéressée à la corrélation des mesures en OCT et d’AV dans l’OMD. Il s’agissait d’une étude longitudinale, cross-sectionnelle portant sur 251 yeux sélectionnés de manière randomisée. L’épaisseur maculaire était mesurée par time-domain optical coherence tomography (TD-OCT) et l’AV à l’aide de l’échelle ETDRS. La corrélation était de 0,52 à l’inclusion et passait de 0,49 à 3,5 mois à 0,38 à 12 mois du traitement par laser focal. De plus, deux sous-groupes de patient montraient une relation paradoxale entre AV et OCT. En effet, on a observé un groupe avec augmentation de l’AV malgré l’augmentation de l’épaisseur maculaire à l’OCT (7–17 %) et un autre avec baisse de l’AV malgré la baisse de l’épaisseur maculaire à l’OCT (18–26 %). Cette étude montre une faible corrélation entre l’AV et l’OCT dans l’OMD, car il peut exister une grande variabilité d’AV pour une même épaisseur à l’OCT avec d’éventuelles relations inverses entre AV et OCT. L’étude SCORE s’est intéressée à cette même corrélation dans l’OM après occlusion veineuse rétinienne centrale et de branche [15]. L’étude a porté sur 665 yeux de manière multicentrique, randomisée associant occlusion veineuse rétinienne centrale (n = 262) et de branche (n = 403). Le coefficient de corrélation entre les mesures d’épaisseur maculaire centrale (effectuée en TD-OCT) et les mesures d’AV (en échelle ETDRS) était de –0,27 pour les OVCR et de –0,28 pour les OBVR. Le modèle de régression linéaire a estimé la baisse d’AV pour 100 µ à l’OCT de 1,7 lettre (p = 0,0007) pour les OVCR et de 1,9 lettre (p < 0,0001) pour les OBVR. Les autres facteurs intervenants étaient la durée de l’OM et la présence de logettes intrarétiniennes. Cette étude confirme la faible corrélation entre l’AV et l’OCT dans les OM post-occlusion veineuse rétinienne. L’OCT est donc un paramètre important mais ne remplace pas la mesure d’AV pour évaluer la fonction visuelle.
La corrélation entre tous les paramètres a été rarement étudiée dans les OM. Nisic et al. ont proposé une étude intéressante sur la corrélation des mesures en OCT avec l’AV en y ajoutant l’examen du fond d’œil au biomicroscope. Cette étude prospective portant sur 90 patients sur 1 an a montré une corrélation positive entre les différents paramètres [16]. Les figures 4-31 à 4-34 présentent de manière multimodale la variation de corrélation entre la sévérité de l’OM et l’AV selon différentes étiologies (diabète, Irvine-Gass, DMLA et OVR).
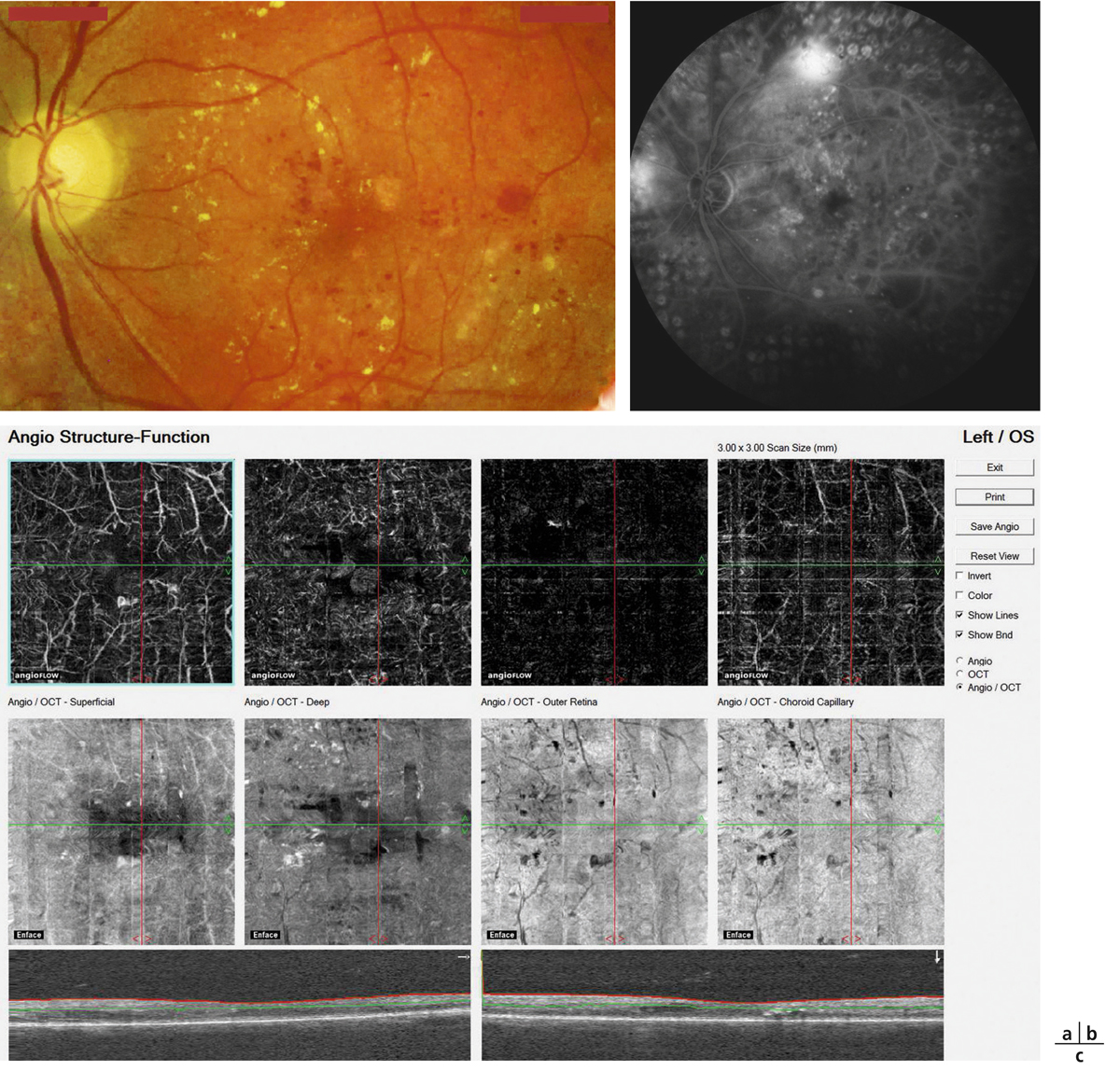
Fig. 4-31 Œdème maculaire diabétique visualisée en rétinographie (a), angiographie à la fluorescéine (b) et angio-OCT (c) avec une AV de 20/50 (64 lettres).
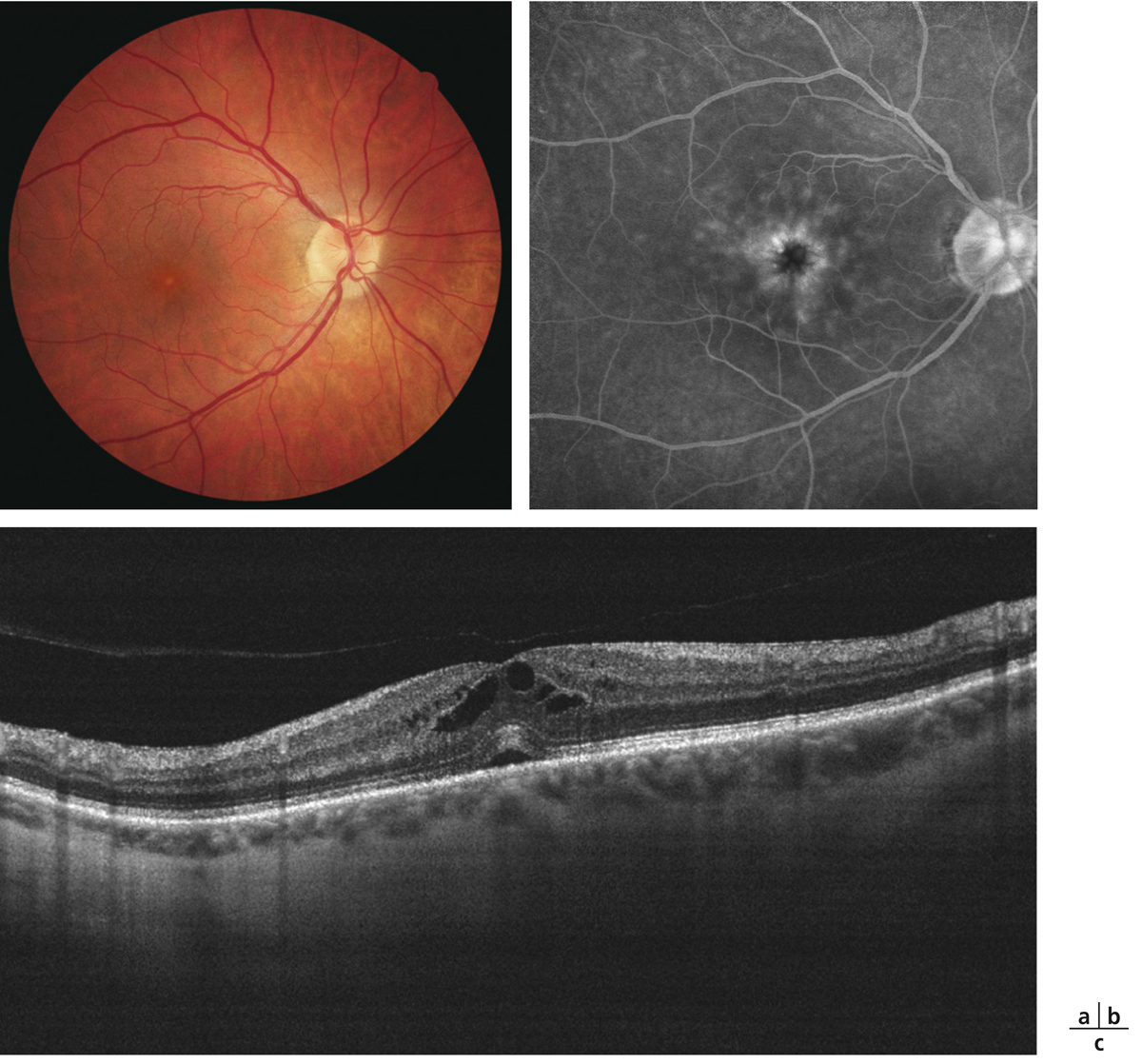
Fig. 4-32 Œdème maculaire sur syndrome d’Irvine-Gass en rétinographie (a), angiographie à la fluorescéine (b) et OCT (c) avec une AV de 20/50 (64 lettres).
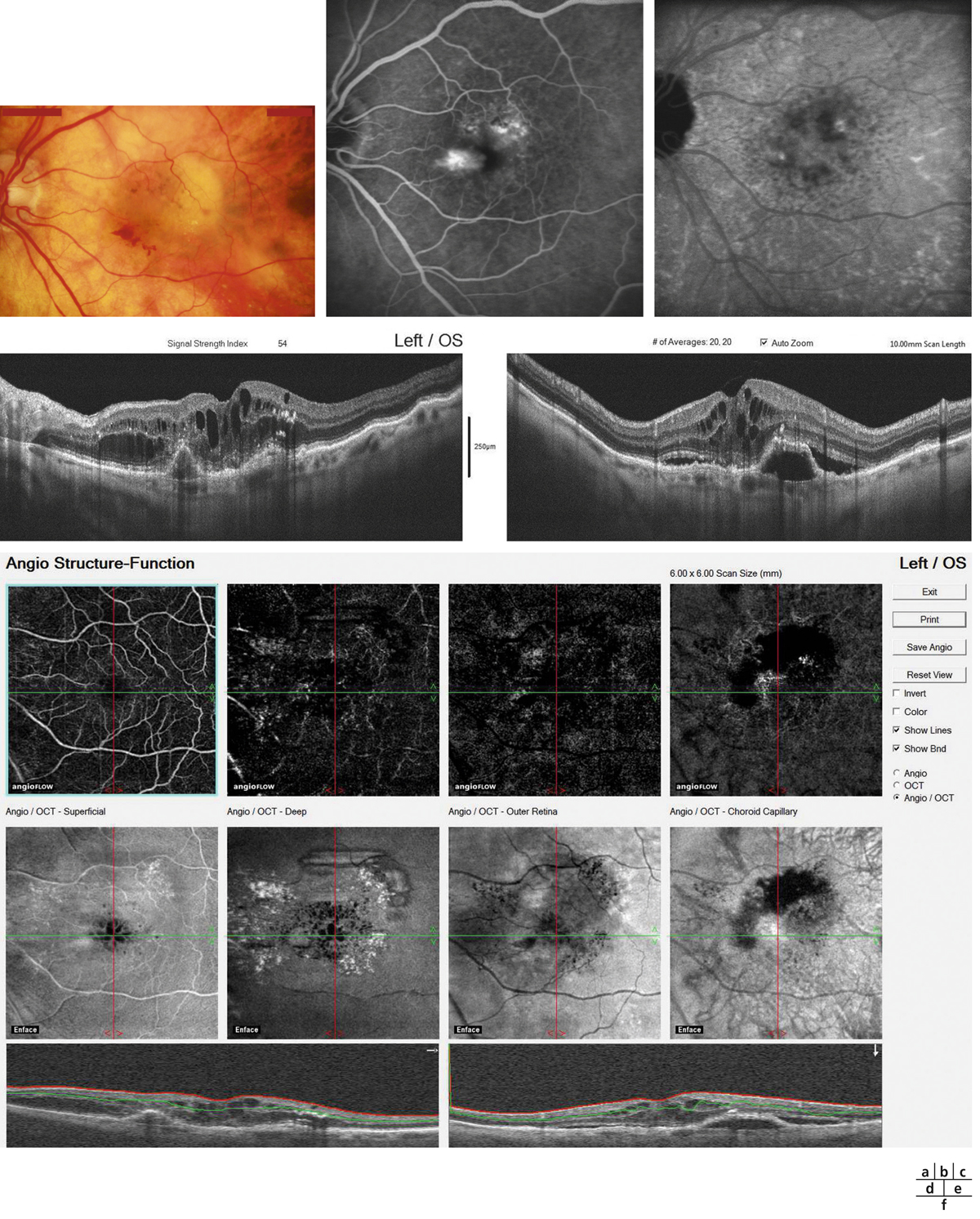
Fig. 4-33 Œdème maculaire sur DMLA néovasculaire avec anastomose choriorétinienne en rétinographie (a), angiographie à la fluorescéine (b), ICG (c), OCT (d, coupe verticale ; e, coupe horizontale) et angio-OCT (f) avec une AV de 20/80 (55 lettres).
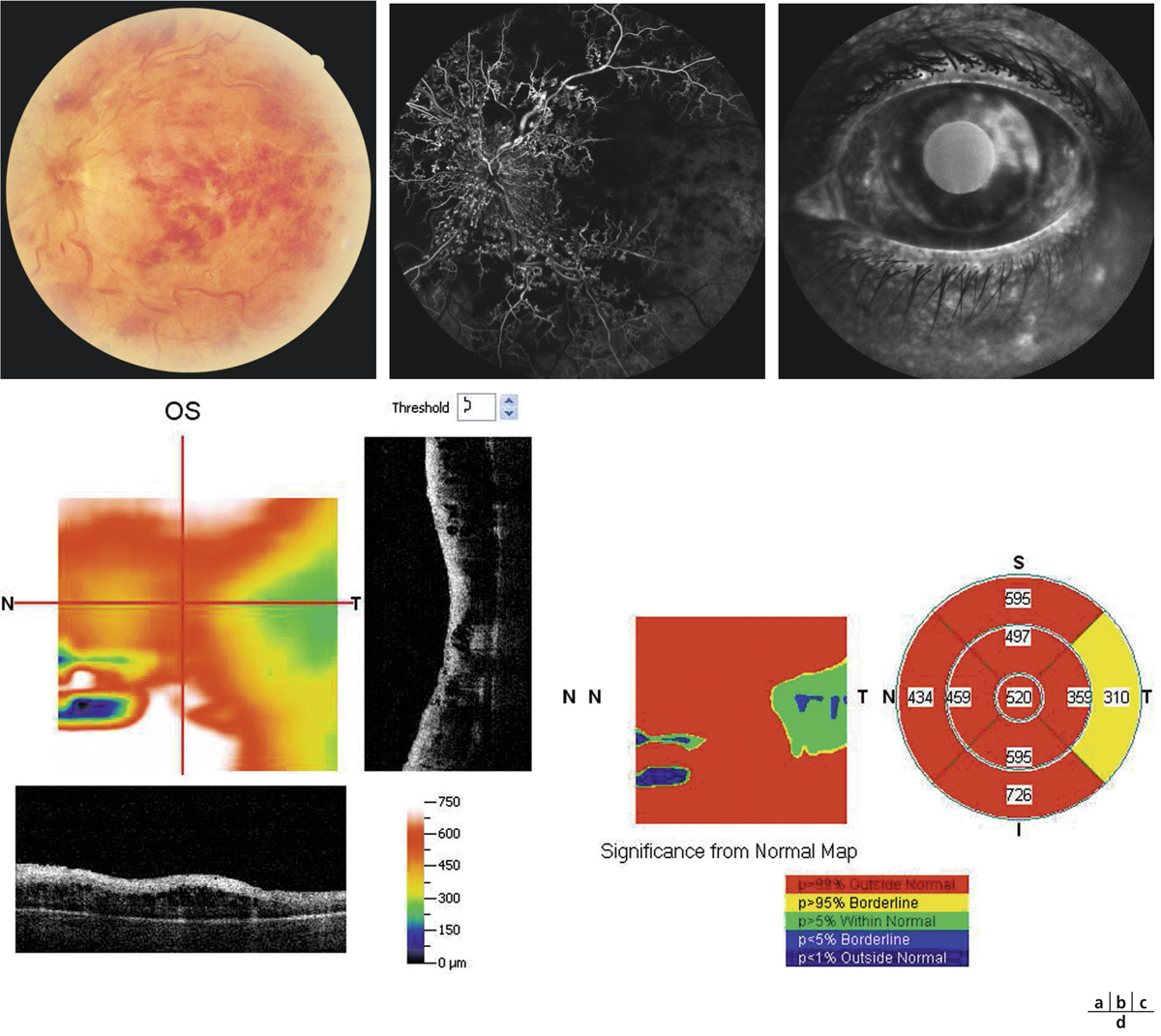
Fig. 4-34 Œdème maculaire sur occlusion veineuse rétinienne ischémique en rétinographie (a), angiographie à la fluorescéine (b), focalisée sur l’iris (c), et OCT (d, coupe et cartographie) avec une AV de 20/50 (64 lettres).
Au vu des différentes études de corrélation, nous pouvons recommander les points suivants pour les études cliniques mais également pour la pratique courante des diagnostics et suivis d’OM :
-
l’acuité visuelle doit être idéalement réalisée à l’aide de l’échelle ETDRS ;
-
l’échelle ETDRS est plus adaptée aux faibles AV (< 20/200) ;
-
la corrélation entre les échelles d’AV Snellen et ETDRS est faible, principalement dans les DMLA néovasculaires ;
-
il convient d’associer des explorations fonctionnelles aux explorations anatomiques ;
-
l’OCT doit être fait systématiquement dans les OM pour obtenir des mesures objectives, précises, fiables et reproductibles ;
-
l’OCT est un paramètre utile mais pas suffisant pour l’exploration des OM.
[1] Ciardella AP, Klancnik J, Schiff W, et al. Intravitreal triamcinolone for the treatment of refractory diabetic macular oedema with hard exudates : an optical coherence tomography study. Br J Ophthalmol 2004 ; 88 : 1131-6.
[2] Funatsu H, Yamashita H, Ikeda T, et al. Vitreous levels of interleukin-6 and vascular endothelial growth factor are releated to diabetic macular edema. Ophthalmology 2008 ; 110 : 1690-6.
[3] Diabetic Retinopathy Clinical Research Network. The relationship between OCT-measured central retinal thickness and visual acuity in diabetic macular edema. Ophthalmology 2007 ; 114 : 525-36.
[4] Kang W, Park Y, Ham DI. The Correlation between fluorescein angiographic and optical coherence tomographic features in clinical significant diabetic macular edema. Am J Ophthalmol 2004 ; 137 : 313-22.
[5] Puech B. Du signe clinique au diagnostic. (Imagerie et exploration de la vision.) BSOF 2012.
[6] Menu JP, Corbe C. Stimulus visuel et différentes fonctions visuelles. Encycl Med Chir (Elsevier, Paris), Ophtalmologie, 21-027-A20. 1991 : p. 26.
[7] Speeg-Schatz C, Zanlonghi X. Acuité visuelle. In : Risse JF, Ed. Techniques d’exploration. Exploration de la Fonction visuelle. Rapport de la Société française d’ophtalmologie. Paris : Masson ; 1999, p. 99-126.
[8] American Academy of ophtalmolgy. Optique clinique, section 3. (Trad. C. Albou-Ganem et al.) Issy-les-Moulineaux : Elsevier Masson ; 2014.
[9] Kaiser PK. Prospective evaluation of visual acuity assessment : a comparison of Snellen versus ETDRS charts in clinical practice. Trans Am Ophthalmol Soc 2009 ; 107 : 311-24.
[10] Falkenstein IA, Cochran DE, Azen SP, et al. Comparison of visual acuity in macular degeneration patients measured with Snellen and Early Treatment Diabetic Retinopathy Study charts. Ophthalmology 2008 ; 115 : 319-23.
[11] Lovie-Kitchin JE. Validity and reliability of visual acuity measurements. Ophthalmic Physiol Opt 1988 ; 8 : 363-70.
[12] Pandit JC. Testing acuity of vision in general practice : reaching recommended standard. BMJ 1994 ; 26 : 1408.
[13] Sheedy JE. Standards for VA measurement. In : Eye care technology forum proceedings. Bethesda, Maryland : NIH ; 1993.
[14] Blackhurst DW, Maguire MG. Reproductibility of refraction and visual acuity measurements under a standard protocol. The macular Photocoagulation Study Group. Retina 1989 ; 9 : 163-9.
[15] Scott IU, VanVeldhuisen PC, Oden NL, et al. SCORE study report 1 : baseline associations between central retinal thickness and visual acuity in patients with retinal vein occlusion. Ophthalmology 2009 ; 116 : 504-12.
[16] Nisic F, Turkovic S, Mavija M, et al. Correlation between the findings of optical coherent retinal tomography (OCT), stereo biomicroscopic images from fundus of an eye and values from visual acuity of diabetic macular edema. Acta Inform Med 2014 ; 22 : 232-6.
C.-J. Pournaras, J.-A. Pournaras
➤ Le débit sanguin rétinien dépend de la pression de perfusion (différence entre la pression dans l’artère centrale de la rétine et la pression intra-oculaire), de la résistance propre des vaisseaux rétiniens et de la viscosité sanguine.
➤ L’apport en oxygène de la rétine dépend de deux sources : les réseaux vasculaire rétinien et choroïdien. La pression partielle en oxygène dans la rétine varie avec la proximité vasculaire et est minimale au niveau des segments internes des photorécepteurs.
➤ L’autorégulation du flux sanguin rétinien maintient constant l’approvisionnement de la rétine en substrats métaboliques et en oxygène. Elle amortit les variations de pression artérielle, de pression intra-oculaire et des besoins métaboliques.
➤ Le seul mécanisme par lequel les vaisseaux rétiniens peuvent exercer cette autorégulation est une variation de diamètre. Différents facteurs sont impliqués dans l’autorégulation, tels que l’oxyde nitrique qui induit une vasodilatation et l’endothéline 1 qui induit une vasoconstriction artériolaire.
➤ Lors des micro-angiopathies ischémiques, la diminution du débit sanguin de la rétine entraîne une hypoxie tissulaire au niveau de la rétine interne et des altérations de la BHR interne, à l’origine d’un œdème rétinien.
Les altérations de la circulation rétinienne, entraînant une baisse du débit, affectent l’apport en oxygène et en substrats métaboliques nécessaires pour entretenir les réactions métaboliques du tissu rétinien.
L’apport approprié en oxygène et en glucose est essentiel pour l’état énergétique assuré par la formation d’adénosine 5'-triphosphate (ATP). Celle-ci est produite au niveau cellulaire, par deux voies métaboliques essentielles : la glycolyse et la phosphorylation oxydative, couplées au cycle de l’oxyde nitrique.
Le débit sanguin rétinien dépend de la pression de perfusion (la pression qui dirige le sang dans le système vasculaire rétinien) et de la résistance vasculaire générée par les vaisseaux rétiniens et la viscosité sanguine.
Le diamètre des vaisseaux de résistance rétinienne est modulé par l’interaction de multiples facteurs systémiques et des mécanismes de contrôle locaux régulant le tonus des péricytes et des cellules musculaires lisses de la paroi des artérioles.
Le flux sanguin rétinien est autorégulé, c’est-à-dire maintenu relativement constant malgré des variations de la pression de perfusion ; l’autorégulation est ainsi définie comme la capacité d’un organe de réguler son propre approvisionnement en fonction de ses besoins métaboliques [1]. Les interactions entre les substances relâchées par les cellules endothéliales vasculaires, les cellules gliales ou les neurones durant les échanges métaboliques, affectent le tonus artériolaire, régulant de cette manière les réponses vasomotrices rétiniennes [2].
L’équation de Krogh-Erlang décrit les relations entre le diamètre d’une artériole et, par conséquent, le débit sanguin et la pression partielle d’oxygène (PO2) locale.
La rétine des mammifères exprime un taux élevé de glycolyse et de production de lactate [3] et, comme dans le système nerveux central, une forte consommation d’oxygène [4, 5]. Ces réactions sont nécessaires pour que la totalité des processus de transport neuronal soit maintenue, en gardant les gradients ioniques nécessaires à l’activité électrique et à la transduction visuelle [6].
Des mesures directes de la PO2 ont été réalisées dans les diverses couches rétiniennes de différentes espèces animales en utilisant des micro-électrodes sensibles à l’oxygène [7–9].
La PO2 proche de l’interface vitréorétinienne est hétérogène [10]. Les profils prérétiniens et transrétiniens de la PO2 indiquent que la diffusion de l’O2, provenant des artérioles, influence la PO2 dans la région juxta-artériolaire prérétinienne et dans les couches rétiniennes internes. À l’opposé, à la fois la PO2 prérétinienne et celle de la rétine interne (à 30 % de profondeur rétinienne) loin des vaisseaux demeurent constantes dans toutes les régions rétiniennes. À l’aide des mesures de PO2 intervasculaires transrétiniennes, il a été observé que la PO2 intrarétinienne diminue graduellement à partir de la surface de la rétine interne et de la choroïde en direction de la rétine moyenne avec une valeur minimum enregistrée à 50 % de profondeur rétinienne [9, 10].
L’ischémie induit des changements du flux sanguin, du métabolisme rétinien et de la distribution de l’oxygène au niveau de la rétine. Après occlusion veineuse expérimentale, une diminution du diamètre artériolaire dans les régions ischémiques se produit suite à des altérations du processus métabolique produisant l’oxyde nitrique rétinien responsable du maintien du tonus vasculaire [11] ou la vasoconstriction artériolaire résultant des interactions entre l’endothéline 1 (ET-1) et le récepteur ETA (endothelin A) de l’endothélium vasculaire [12].
Des régions rétiniennes affectées par une occlusion de branche veineuse aiguë [13], ou chez des rats diabétiques [14], montrent une hypoxie tissulaire de la rétine interne. Elle est le résultat de la diminution de l’approvisionnement sanguin. Les mesures de la PO2 sont aussi pertinentes que les mesures du flux sanguin lui-même. De même, la PO2 tissulaire, en accord avec la loi de diffusion de Fick, est étroitement associée au diamètre vasculaire et à la distribution (c’est-à-dire la distance d’un vaisseau) dans une région de mesure [5].
Un débit sanguin réduit et une imprégnation des parois vasculaires en angiographie à la fluorescence sont associés à une ischémie vasculaire sévère chez des patients présentant une rétinopathie diabétique proliférante [15].
Il est bien établi qu’en présence d’une néovascularisation rétinienne et d’un œdème maculaire diabétique, l’hypoxie induit fortement l’expression du VEGF par la stabilisation du facteur de transcription hypoxia-inducible factor 1 (HIF-1) [16, 17]. Dans la rétine ischémique, des taux élevés de HIF-1 précèdent la surexpression de VEGF qui est corrélée à la formation de néovaisseaux. Des taux élevés de VEGF ont été retrouvés dans une rétine ischémique de singe affectée par une occlusion de branche veineuse [18] mais également dans le sang [19] et le vitré de patients atteints d’ischémie rétinienne [20, 21]. Ces résultats indiquent que l’expression du VEGF est étroitement corrélée à une angiogenèse non contrôlée.
De plus, la régulation du flux sanguin rétinien est altérée durant l’évolution des micro-angiopathies ischémiques. L’atteinte de la paroi vasculaire associée à des altérations des propriétés rhéologiques du sang pourrait affecter la capacité de la rétine diabétique à réguler son flux sanguin. Les patients atteints de rétinopathie diabétique proliférative présentent une altération de la régulation du débit sanguin de la rétine, soit dans des conditions rétiniennes hypoxiques [22] ou lors d’une augmentation de la concentration en oxygène de l’air inhalé [23, 24].
La capacité de la circulation rétinienne à répondre à des changements de la pression de perfusion moyenne (PPm) oculaire, diminuée par une augmentation de la pression intra-oculaire ou augmentée lors de l’exercice physique, est altérée chez le patient diabétique [25]. Une altération de l’autorégulation des artérioles rétiniennes corrélée à la sévérité de la rétinopathie diabétique a été montrée à l’aide du retinal vessel analyser ou RVA (fig. 4-35) [26].
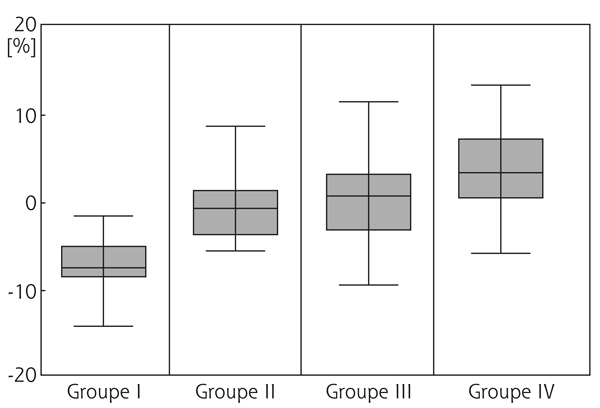
Fig. 4-35 Une altération de l’autorégulation des artérioles rétiniennes corrélée à la sévérité de la rétinopathie diabétique (RD) a été montrée à l’aide du retinal vessel analyser (RVA).
En réponse à une augmentation de la pression sanguine similaire, le groupe I sans RD a présenté une vasoconstriction physiologique de − 7,6 % + 4,1, et le groupe II avec RD modérée une vasoconstriction de − 0,85 % + 4,3. En revanche, dans le groupe III avec RD non proliférante sévère et le groupe IV avec une RD proliférante, on observe une vasodilatation pathologique respectivement de + 0,44 % + 5,9 et + 3,2 % + 5,9.
(Source : Blum M, et al. The myogenic response of retinal arterioles in diabetic retinopathy. Ophthalmologe 2006 ; 103 : 209-13. Avec l’autorisation de l’éditeur.)
La BHR interne contrôle l’échange entre la circulation sanguine et le parenchyme rétinien.
La BHR interne est définie par l’unité neurovasculaire composée de l’endothélium, des cellules de la paroi vasculaire (péricytes et cellules vasculaires lisses) et de la glie périvasculaire (astrocytes, cellules de Müller, microglie). Elle maintient l’homéostasie rétinienne en régulant le flux sanguin des vaisseaux intrarétiniens et l’activité synaptique [27, 28].
-
Endothélium. Le réseau complexe de jonctions serrées et jonctions adhérentes entre les cellules endothéliales capillaires, l’absence de fenestrations et la rareté relative de vésicules de transport transcellulaire contribuent à l’étanchéité de la BHR interne. Des systèmes de transport spécifiques assurent la sélectivité de la barrière [29].
-
Péricytes. Le contact entre cellules endothéliales et péricytes à travers une lame basale commune serait déterminant pour leur renouvellement ainsi que pour l’étanchéité de la BHR interne. Les péricytes, eux-mêmes dépourvus de jonctions serrées mais contenant un nombre élevé de vésicules de transport (passage transcellulaire), sont considérés comme modulateurs de la perméabilité. Ainsi, la perte de péricytes est corrélée à une augmentation de la perméabilité de la barrière et une déficience de couverture péricytaire entraîne une réduction des protéines des jonctions serrées. De plus, les péricytes confèrent un support structural dynamique à l’endothélium par la présence de protéines contractiles, des canaux ioniques et des récepteurs de ligands vaso-actifs, permettant une contraction ou une dilatation des capillaires qu’ils entourent.
-
Cellules gliales. La paroi des capillaires rétiniens, comme celui des capillaires du cerveau, n’a pas d’enveloppe conjonctive mais est en contact avec des prolongements de cellules gliales. Par sa position à l’interface entre vaisseau et tissu neural, la glie ne sert pas seulement comme support structural mais joue un rôle essentiel dans le couplage métabolique de l’activité neuronale au flux sanguin. Elle est engagée dans la régulation des barrières en sécrétant des facteurs humoraux qui affectent la qualité de la barrière au niveau des cellules endothéliales. Une altération gliale est une cause de déficience de la barrière. De plus, à l’intérieur de la rétine, l’homéostasie rétinienne et le drainage de l’eau sont assurés par les cellules gliales qui sont dotées d’un grand nombre de molécules d’aquaporines. Formant des pores, ces protéines intramembranaires régulent l’évacuation de l’eau vers l’espace extracellulaire lors de l’activité neuronale [30, 31]. La fonction d’évacuation d’eau n’est plus assurée dans les situations ischémiques (occlusion artérielle rétinienne, diabète, hypertension) menant à la formation d’œdème [32].
Dans les organes neuraux, y compris la rétine, le VEGF est produit par les neurones, les cellules gliales, l’épithélium pigmentaire de la rétine (EPR) et les cellules vasculaires. Il est le principal inducteur d’une perméabilité vasculaire accrue. Son expression augmentée lors des conditions d’hypoxie amène à : la formation de fenestrations endothéliales ; une perméabilité élevée à l’égard de différentes substances, y compris le sucrose ; une diminution de la résistance transendothéliale électrique et, au niveau des jonctions serrées, une perte des composants protéiques. Ce processus est réversible, indiquant une plasticité marquée des vaisseaux.
Dans les pathologies liées à l’ischémie et l’hypoxie, l’augmentation de la perméabilité de la BHR interne est provoquée également par un certain nombre de facteurs liés à l’inflammation, notamment cytokines, facteurs angiogéniques, un stress oxydatif et des concentrations élevées d’oxyde nitrique [33].
L’hypoxie induit une hyperperméabilité paracellulaire au niveau des capillaires rétiniens secondaire à l’altération de la synthèse des protéines des jonctions serrées et d’une perturbation de leur arrangement spatial. En effet, sous conditions d’hypoxie, la répartition des protéines occludine, zonula occludens 1 et 2, le long des membranes des cellules endothéliales serait d’aspect discontinu, liée au déplacement de leur expression vers le cytoplasme et le noyau de la cellule (eFig. 4-1 ). Ces modifications entraîneraient une augmentation de la perméabilité paracellulaire [34].
). Ces modifications entraîneraient une augmentation de la perméabilité paracellulaire [34].
Des modifications de la distribution d’occludine au niveau des jonctions serrées de l’endothélium vasculaire des vaisseaux de la BHR interne chez le rat normal ex vivo (eFig. 4-2a ) et suite à une occlusion expérimentale d’une branche veineuse (eFig. 4-2b et c) ont été démontrées.
) et suite à une occlusion expérimentale d’une branche veineuse (eFig. 4-2b et c) ont été démontrées.
La meilleure preuve du rôle du VEGF et des facteurs inflammatoires, dans le phénomène d’hyperperméabilité capillaire et d’œdème maculaire et la néovascularisation caractérisant plusieurs maladies rétiniennes, est l’effet inhibiteur du traitement anti-VEGF et des inhibiteurs de l’inflammation sur ces altérations.
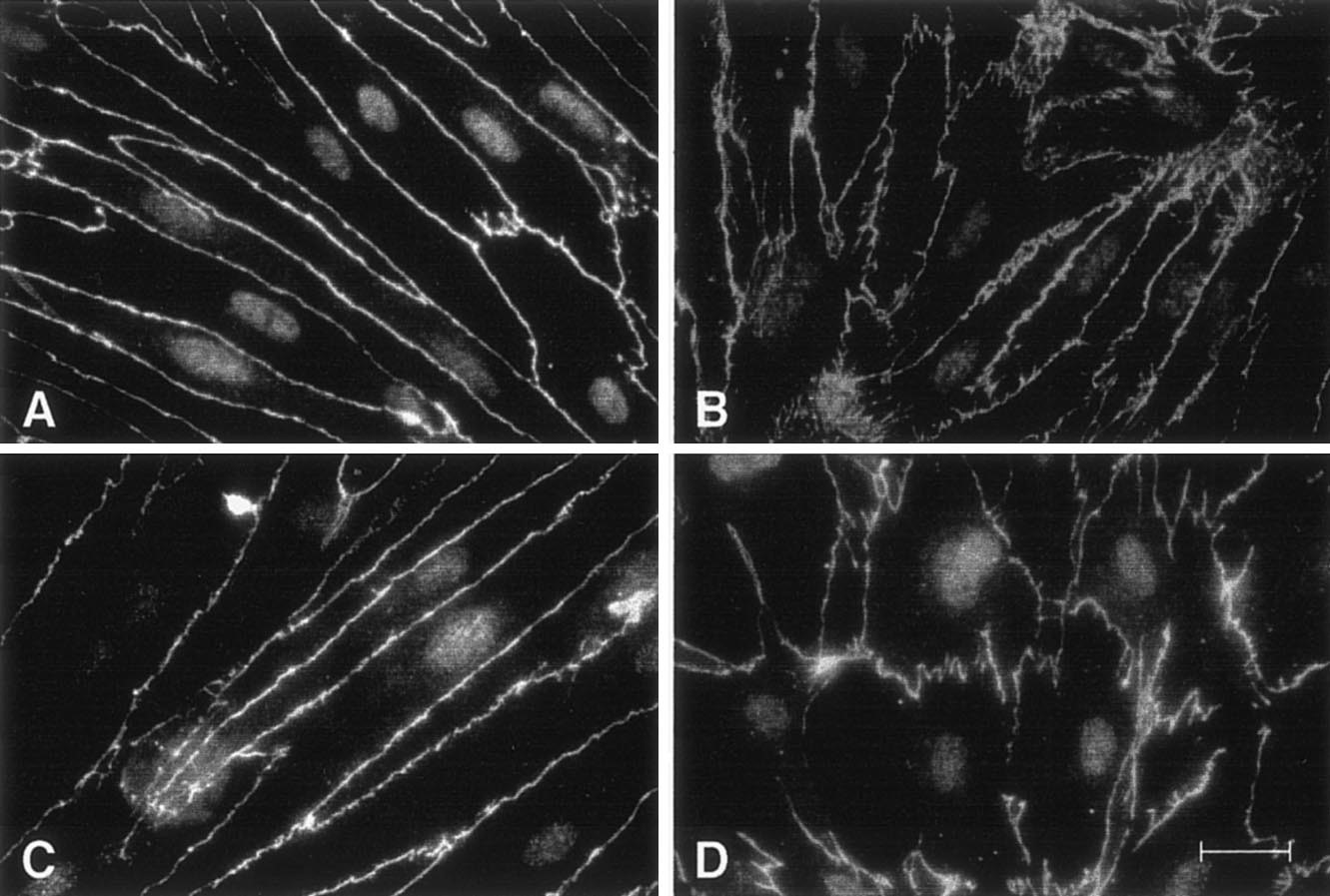
eFig. 4-1 Effet de l’hypoxie et du VEGF en présence d’acide α-lipoïque (LA) sur la distribution intercellulaire de la protéine zonula occludens 1 (ZO-1).
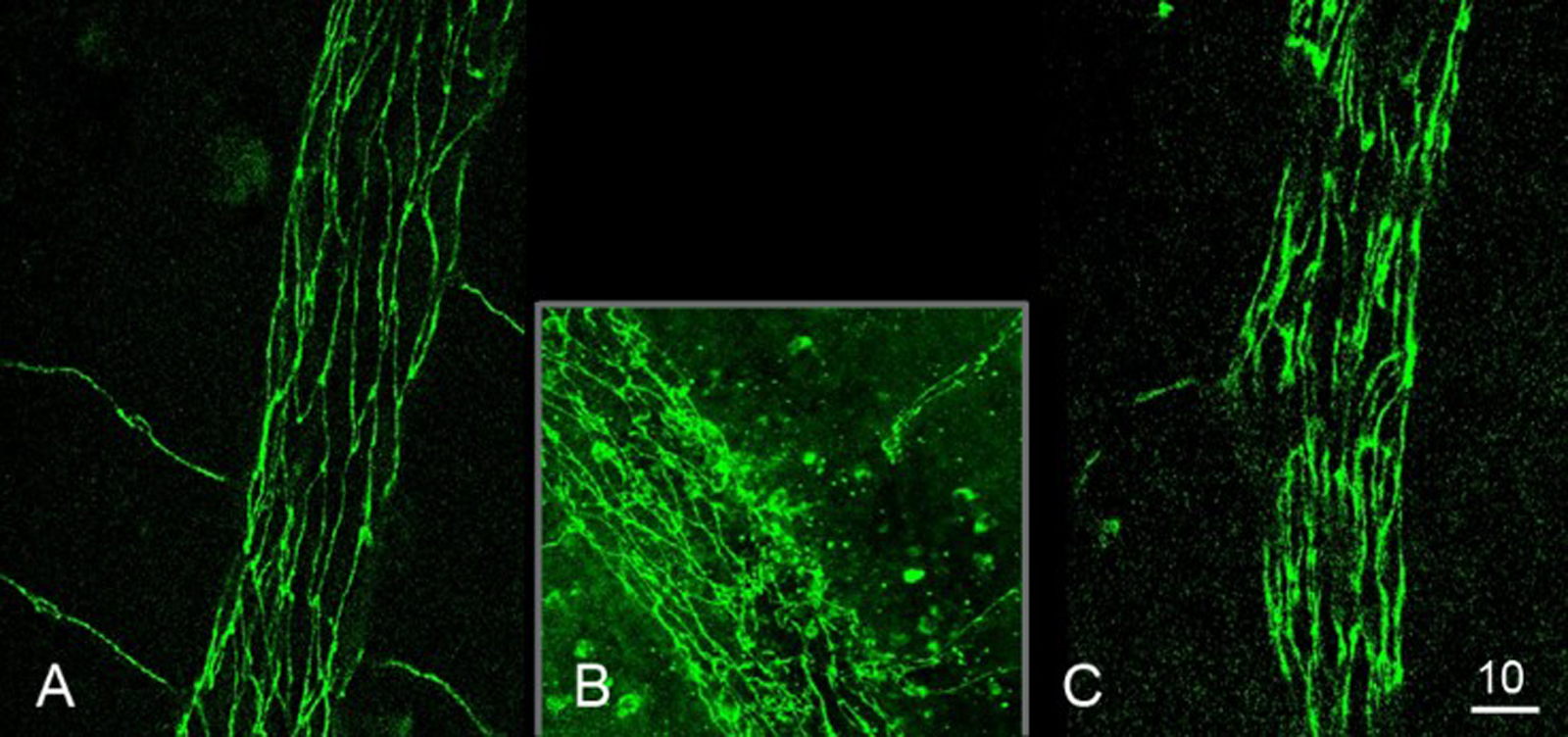
eFig. 4-2 Mise en évidence de l’occludine au niveau de l’endothélium de la BHR interne chez le rat normal et suite à une occlusion expérimentale d’une branche veineuse.
La loi de Starling décrit des échanges d’eau dans un état stationnaire entre le compartiment vasculaire et le tissu extracellulaire [35]. La pression hydrostatique (P) dans le vaisseau dirige l’eau en direction du tissu et est opposée aux différences de pression oncotique (osmotique) entre le sang et le tissu. La loi de Starling établit que, pour un équilibre dans les échanges de fluide entre les vaisseaux et le tissu, les gradients de pression hydrostatique et oncotique doivent être égaux et opposés.
Les artérioles rétiniennes servent de vaisseaux de résistance et contrôlent la pression hydrostatique des capillaires en aval. Des artérioles dilatées, en cas d’altération de l’autorégulation, offrent moins de résistance ; en conséquence, le flux sanguin et la pression hydrostatique sont augmentés dans les capillaires en aval et dans les veinules, où la pression hydrostatique élevée dilate ces vaisseaux à paroi fine. Une augmentation de la pression hydrostatique dans les capillaires et les veinules dirige l’eau dans le tissu et crée l’œdème extracellulaire.
L’augmentation de l’oxygène dans l’air inhalé ou la régression de l’hypoxie rétinienne obtenue par la photocoagulation au laser entraînent une vasoconstriction rétinienne qui diminue la pression hydrostatique dans les capillaires et les veinules. Cette réduction fait baisser de ce fait d’une part, la fuite à travers la paroi vasculaire et d’autre part, la formation de l’œdème [36]. Par ailleurs l’oblitération par le laser des segments pathologiques des capillaires réduit l’œdème extracellulaire. L’augmentation de l’oxygénation de la rétine interne diminue l’expression du VEGF et la perméabilité vasculaire.
L’autorégulation du flux sanguin rétinien maintient relativement constant l’approvisionnement de la rétine en substrats métaboliques et en oxygène. Pendant l’évolution des micro-angiopathies ischémiques, la diminution du débit sanguin de la rétine entraîne une hypoxie tissulaire au niveau de la rétine interne et des altérations de la BHR interne. Les altérations hypoxiques des cellules neuronales et gliales de la rétine interne et l’œdème extracellulaire lié à la perméabilité pathologique de la BHR interne aboutissent à la formation de l’œdème maculaire. La photocoagulation, les inhibiteurs des facteurs vasoprolifératifs et le contrôle des facteurs systémiques, en réduisant l’hypoxie et rétablissant la régulation du débit, peuvent réduire l’œdème rétinien.
[1] Guyton AC, Jones CE, Coleman TG. Circulatory physiology : cardiac output and its regulation. Philadelphia : W.B. Saunders Company ; 1973.
[2] Pournaras CJ, Rungger-Brandle E, Riva CE, Hardarson SH, Stefansson E. Regulation of retinal blood flow in health and disease. Prog Retin Eye Res 2008 ; 27 : 284-330.
[3] Winkler BS. Glycolytic and oxidative metabolism in relation to retinal function. J Gen Physiol 1981 ; 77 : 667-92.
[4] Weiter JJ, Zuckerman R. The influence of the photoreceptor-RPE complex on the inner retina. An explanation for the beneficial effects of photocoagulation. Ophthalmology 1980 ; 87 : 1133-9.
[5] Tsacopoulos M. Various aspects of the existing relationship between retinal neuronal function, oxidative metabolism and the constant O2 supply by the microcirculation. Klin Monatsbl Augenheilkd 1985 ; 186 : 477-9.
[6] Penn RD, Hagins WA. Kinetics of the photocurrent of retinal rods. Biophys J 1972 ; 12 : 1073-94.
[7] Alder VA, Cringle SJ, Constable IJ. The retinal oxygen profile in cats. Invest Ophthalmol Vis Sci 1983 ; 24 : 30-6.
[8] Linsenmeier RA. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J Gen Physiol 1986 ; 88 : 521-42.
[9] Pournaras CJ, Riva CE, Tsacopoulos M, Strommer K. Diffusion of O2 in the retina of anesthetized miniature pigs in normoxia and hyperoxia. Exp Eye Res 1989 ; 49 : 347-60.
[10] Riva CE, Pournaras CJ, Tsacopoulos M. Regulation of local oxygen tension and blood flow in the inner retina during hyperoxia. J Appl Physiol 1986 ; 61 : 592-8.
[11] Donati G, Pournaras CJ, Pizzolato GP, Tsacopoulos M. Decreased nitric oxide production accounts for secondary arteriolar constriction after retinal branch vein occlusion. Invest Ophthalmol Vis Sci 1997 ; 38 : 1450-7.
[12] Stangos AN, Petropoulos IK, Pournaras JA, et al. The vasodilatory effect of juxta-arteriolar microinjection of endothelin A receptor inhibitor in healthy and acute branch retinal vein occlusion minipig retinas. Invest Ophthalmol Vis Sci 2010 ; 51 : 2185-90.
[13] Pournaras CJ, Tsacopoulos M, Strommer K, et al. Experimental retinal branch vein occlusion in miniature pigs induces local tissue hypoxia and vasoproliferative microangiopathy. Ophthalmology 1990 ; 97 : 1321-8.
[14] Linsenmeier RA, Braun RD, McRipley MA, et al. Retinal hypoxia in long-term diabetic cats. Invest Ophthalmol Vis Sci 1998 ; 39 : 1647-57.
[15] Grunwald JE, Brucker AJ, Grunwald SE, Riva CE. Retinal hemodynamics in proliferative diabetic retinopathy. A laser Doppler velocimetry study. Invest Ophthalmol Vis Sci 1993 ; 34 : 66-71.
[16] Semenza GL. HIF-1 : mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 2000 ; 88 : 1474-80.
[17] Arjamaa O, Nikinmaa M. Oxygen-dependent diseases in the retina : role of hypoxia-inducible factors. Exp Eye Res 2006 ; 83 : 473-83.
[18] Miller JW, Adamis AP, Shima DT, et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am J Pathol 1994 ; 145 : 574-84.
[19] Lip PL, Belgore F, Blann AD, et al. Plasma VEGF and soluble VEGF receptor FLT-1 in proliferative retinopathy : relationship to endothelial dysfunction and laser treatment. Invest Ophthalmol Vis Sci 2000 ; 41 : 2115-9.
[20] Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994 ; 331 : 1480-7.
[21] Augustin AJ, Keller A, Koch F, et al. Effect of retinal coagulation status on oxidative metabolite and VEGF in 208 patients with proliferative diabetic retinopathy. Klin Monatsbl Augenheilkd 2001 ; 218 : 89-94.
[22] Fallon TJ, Maxwell D, Kohner EM. Retinal vascular autoregulation in conditions of hyperoxia and hypoxia using the blue field entoptic phenomenon. Ophthalmology 1985 ; 92 : 701-5.
[23] Grunwald JE, Riva CE, Brucker AJ, et al. Altered retinal vascular response to 100 % oxygen breathing in diabetes mellitus. Ophthalmology 1984 ; 91 : 1447-52.
[24] Gilmore ED, Hudson C, Nrusimhadevara RK, et al. Retinal arteriolar diameter, blood velocity, and blood flow response to an isocapnic hyperoxic provocation in early sight-threatening diabetic retinopathy. Invest Ophthalmol Vis Sci 2007 ; 48 : 1744-50.
[25] Sinclair SH, Grunwald JE, Riva CE, et al. Retinal vascular autoregulation in diabetes mellitus. Ophthalmology 1982 ; 89 : 748-50.
[26] Blum M, Pils C, Muller UA, Strobel J. The myogenic response of retinal arterioles in diabetic retinopathy. Ophthalmologe 2006 ; 103 : 209-13.
[27] Ruhrberg C, Bautch V. Neurovascular development and links to disease. Cell Mol Life Sci 2013 ; 70: 1675-84.
[28] Vallon M, Chang J, Zhang H, Kuo C. Developmental and pathological angiogenesis in the central nervous system. Cell Mol Life Sci 2014 ; 71 : 3489-506.
[29] Gavard J. Endothelial permeability and VE-cadherin : a wacky comradeship. Cell Adh Migr 2014 ; 8 : 158-64.
[30] Aijaz S, Balda MS, Matter K. Tight junctions : molecular architecture and function. Int Rev Cytol 2006 ; 248 : 261-98.
[31] Verkman AS. Aquaporins in clinical medicine. Annu Rev Med 2012 ; 63 : 303-16.
[32] Bringmann A, Wiedemann P. Muller glial cells in retinal disease. Ophthalmologica 2012 ; 227 : 1-19.
[33] Kaur C, Foulds WS, Ling EA. Blood-retinal barrier in hypoxic ischaemic conditions : basic concepts, clinical features and management. Prog Retin Eye Res 2008 ; 27 : 622-47.
[34] Fischer S, Wiesnet M, Marti HH, et al. Simultaneous activation of several second messengers in hypoxia-induced hyperpermeability of brain derived endothelial cells. J Cell Physiol 2004 ; 198 : 359-69.
[35] Charm SE, Kurlard GS. Starling hypothesis (law). Blood flow and microcirculation. New York : John Wileg and Sons ; 1974.
[36] Stefansson E. Oxygen and diabetic eye disease. Graefes Arch Clin Exp Ophthalmol 1990 ; 228 : 120-3.
- Chapitre 4
Physiopathologie générale des œdèmes maculaires- 1 - Introduction
- 2 - Barrière hémato-rétinienne interne
- 3 - Physiopathogénie de l’œdème maculaire : la barrière hémato-rétinienne externe
- 4 - Mécanismes d’homéostasie et de cohésion de la rétine (rétine normale et altérations en cas d’œdème maculaire)
- 5 - Conséquences de l’œdème sur la rétine
- 6 - Vascular endothelial growth factor (VEGF) et autres cytokines : rôles dans l’œdème maculaire
- 7 - Microglie et notion de para-inflammation dans l’œdème maculaire
- 8 - Rôle du vitré dans l’œdème maculaire
- 9 - Œdème maculaire et vision
- 10 - Flux sanguin et oxygénation rétinienne : liens avec l’œdème maculaire